
Dr. Andrew McArthur is a Professor and the David Braley Chair in Computational Biology in the Michael G. DeGroote Institute for Infectious Disease Research, David Braley Centre for Antibiotic Discovery, and Department of Biochemistry & Biomedical Sciences in the Faculty of Health Sciences at McMaster University. He has had a 25+ year research career in the United States and Canada, including postdoctoral experience at the National Museum of Natural History and NIH-funded faculty positions at the Marine Biological Laboratory (Woods Hole, MA) and Brown University, where he led the computational biology of sequencing the genome of the diarrheal pathogen Giardia intestinalis, plus 10 years experience in the private sector. He was also Director of McMaster’s Biomedical Discovery & Commercialization Program 2019-2024, providing business and drug discovery cross training.
![]() The McArthur laboratory’s research program spans complex informatics approaches to the genomic surveillance of microbial drug resistance, development of biological databases, next generation sequencing for genome assembly and molecular epidemiology, automated literature curation approaches, and controlled vocabularies for biological knowledge integration. A major part of our research program involves our Comprehensive Antibiotic Resistance Database (CARD) which provides data standardization tools for genomic surveillance of drug resistant bacterial pathogens. CARD is now the most widely used database for molecular surveillance of antimicrobial resistance across the globe, with over 8000 citations (increasing by ~60 citations per month), over 4 million page views, and use of its algorithms to analyze the genome of a drug resistant infection once every 4 minutes. In addition, the McArthur Lab had a leadership role in genomic surveillance and bioinformatics methods development to address the SARS-CoV-2 pandemic within Canada, including development of the national Illumina sequencing bioinformatics workflow, genomic confirmation of isolation of the live virus, and provisioning SARS-CoV-2 genome sequence data.
The McArthur laboratory’s research program spans complex informatics approaches to the genomic surveillance of microbial drug resistance, development of biological databases, next generation sequencing for genome assembly and molecular epidemiology, automated literature curation approaches, and controlled vocabularies for biological knowledge integration. A major part of our research program involves our Comprehensive Antibiotic Resistance Database (CARD) which provides data standardization tools for genomic surveillance of drug resistant bacterial pathogens. CARD is now the most widely used database for molecular surveillance of antimicrobial resistance across the globe, with over 8000 citations (increasing by ~60 citations per month), over 4 million page views, and use of its algorithms to analyze the genome of a drug resistant infection once every 4 minutes. In addition, the McArthur Lab had a leadership role in genomic surveillance and bioinformatics methods development to address the SARS-CoV-2 pandemic within Canada, including development of the national Illumina sequencing bioinformatics workflow, genomic confirmation of isolation of the live virus, and provisioning SARS-CoV-2 genome sequence data.
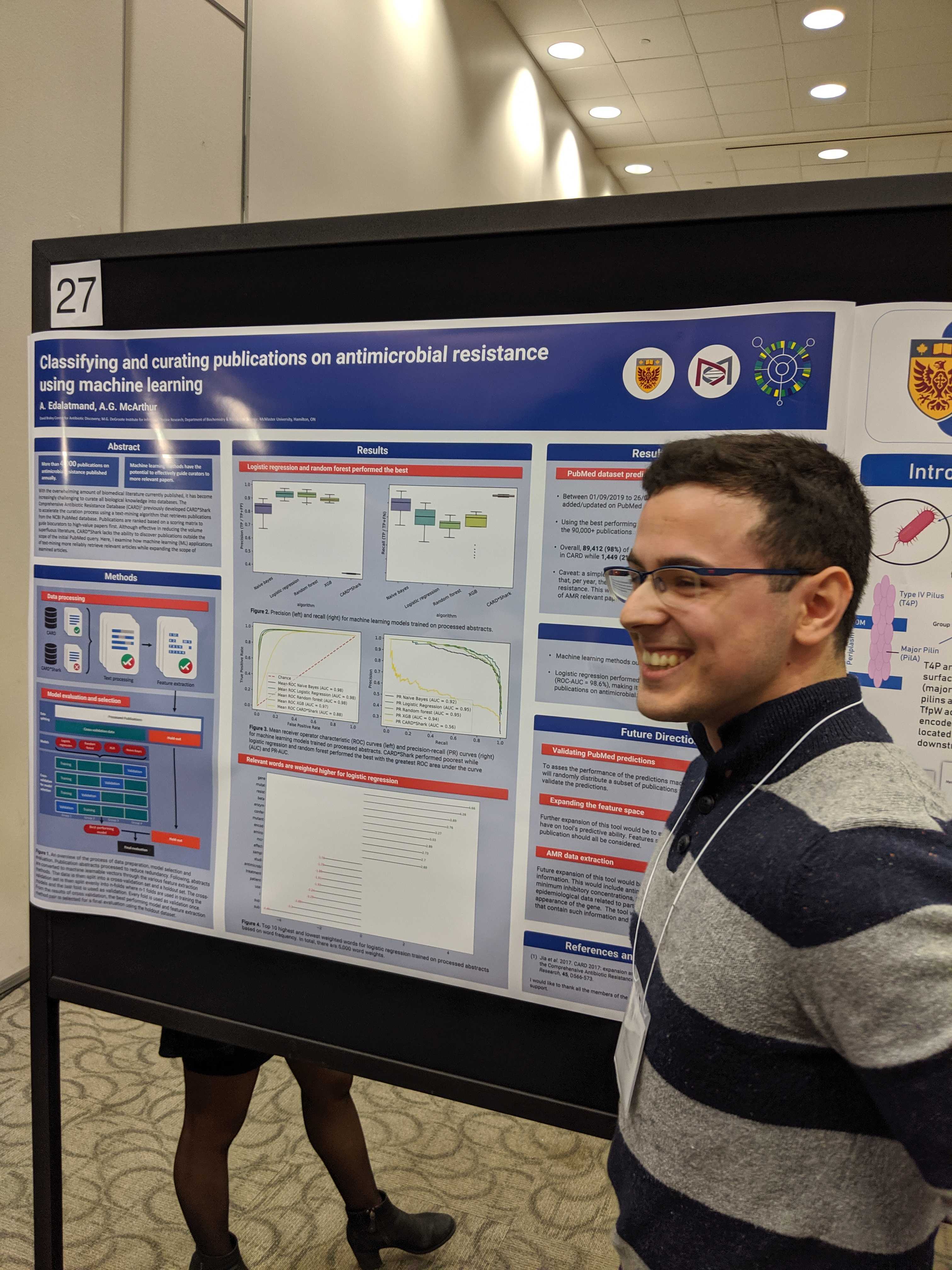 Our research can be broken down into three themes: biocuration, bioinformatics, and analytics. The lab’s biocuration work involves the design of biological databases, such as the Broad Street schema underlying CARD, application of controlled vocabularies for standardization of antimicrobial resistance data, and translation of scientific knowledge into digital algorithms. This work is based on expert knowledge frameworks and personnel, but we are increasingly researching the use of machine learning and natural language processing to develop better data sets to help combat antimicrobial resistance, track movement of resistance genes, and guide drug development. Our bioinformatics work involves development and application of algorithms for prediction of antimicrobial resistance genes and mutations from DNA sequencing or proteomics data, with our premier tool being the widely used Resistance Gene Identifier. Since 2021, we have developed new RGI algorithms for more reliable annotation of antimicrobial resistance genes in complex metagenomics data, providing the first tools for complete resistome prediction for microbiomes, including k-mer tools for pathogen or plasmid identification. An additional component of our bioinformatics work involves collaborations, evolutionary biology, phylogenetics, and genome annotation to better understand biosynthesis of antibiotics, discovery of antimicrobial resistance adjuvants, or bacterial virulence mechanisms. A by-product of this work is application of genomic techniques directly to clinical questions, best exemplified by a recent paper describing Salmonella typhimurium bacteremia in a traveler. Our analytics work is our newest branch of research and involves machine learning to predict complexity, such as using logistic regression to predict antimicrobial resistance phenotype from raw genome sequences or transmission patterns of SARS-CoV-2 using tens of thousands of genome sequences. This has expanded to use of transformer natural language processing to mine the literature for epidemiological patterns for antimicrobial resistance genes.
Our research can be broken down into three themes: biocuration, bioinformatics, and analytics. The lab’s biocuration work involves the design of biological databases, such as the Broad Street schema underlying CARD, application of controlled vocabularies for standardization of antimicrobial resistance data, and translation of scientific knowledge into digital algorithms. This work is based on expert knowledge frameworks and personnel, but we are increasingly researching the use of machine learning and natural language processing to develop better data sets to help combat antimicrobial resistance, track movement of resistance genes, and guide drug development. Our bioinformatics work involves development and application of algorithms for prediction of antimicrobial resistance genes and mutations from DNA sequencing or proteomics data, with our premier tool being the widely used Resistance Gene Identifier. Since 2021, we have developed new RGI algorithms for more reliable annotation of antimicrobial resistance genes in complex metagenomics data, providing the first tools for complete resistome prediction for microbiomes, including k-mer tools for pathogen or plasmid identification. An additional component of our bioinformatics work involves collaborations, evolutionary biology, phylogenetics, and genome annotation to better understand biosynthesis of antibiotics, discovery of antimicrobial resistance adjuvants, or bacterial virulence mechanisms. A by-product of this work is application of genomic techniques directly to clinical questions, best exemplified by a recent paper describing Salmonella typhimurium bacteremia in a traveler. Our analytics work is our newest branch of research and involves machine learning to predict complexity, such as using logistic regression to predict antimicrobial resistance phenotype from raw genome sequences or transmission patterns of SARS-CoV-2 using tens of thousands of genome sequences. This has expanded to use of transformer natural language processing to mine the literature for epidemiological patterns for antimicrobial resistance genes.
See a list of our latest publications!
Significant Contributions
 Genome and Transcriptome of the parasite Giardia lamblia. Giardia is an evolutionary enigma at the cellular transition from prokaryote to eukaryote, but also an important infectious agent and major cause of the world’s diarrheal burden. Dr. McArthur was the ~12 Mbp genome project’s lead bioinformatician, developing sequencing pipelines, shotgun sequence assembly algorithms, genome annotation tools, and its online database. Simultaneous to the genome sequencing effort, he established his own high-throughput genome-wide gene expression laboratory to understand Giardia functional biology. Publication of the complete genome (Morrison et al. 2007. Science, 317, 1921-1926) provided evidence for early evolution of introns, rampant lateral gene transfer, loss of mitochondria, and a diversity of surface proteins important for host-parasite interactions. Transcriptome analysis of Giardia’s complete life cycle (Birkeland et al. 2010. Mol. Biochem. Parasit., 174, 62-65) provided dynamic context to interpretation of its genome, illustrating the fundamental difference in transcriptome between cyst and trophozoite, and highlighted genes important for encystation and excystation.
Genome and Transcriptome of the parasite Giardia lamblia. Giardia is an evolutionary enigma at the cellular transition from prokaryote to eukaryote, but also an important infectious agent and major cause of the world’s diarrheal burden. Dr. McArthur was the ~12 Mbp genome project’s lead bioinformatician, developing sequencing pipelines, shotgun sequence assembly algorithms, genome annotation tools, and its online database. Simultaneous to the genome sequencing effort, he established his own high-throughput genome-wide gene expression laboratory to understand Giardia functional biology. Publication of the complete genome (Morrison et al. 2007. Science, 317, 1921-1926) provided evidence for early evolution of introns, rampant lateral gene transfer, loss of mitochondria, and a diversity of surface proteins important for host-parasite interactions. Transcriptome analysis of Giardia’s complete life cycle (Birkeland et al. 2010. Mol. Biochem. Parasit., 174, 62-65) provided dynamic context to interpretation of its genome, illustrating the fundamental difference in transcriptome between cyst and trophozoite, and highlighted genes important for encystation and excystation.
 Antimicrobial Resistance (AMR) Analytics. In 2014, Dr. McArthur initiated a new research program focused on the problem of antimicrobial resistance, building novel bioinformatics resources in preparation for the approaching transition to routine clinical and environmental DNA sequencing of important bacterial pathogens. Of particular impact has been the construction of the Comprehensive Antibiotic Resistance Database (latest publication: Alcock et al. 2023. Nucleic Acids Res. 51, D690-D699), a curated resource providing reference DNA and protein sequences, detection models and bioinformatics tools on the molecular basis of bacterial antimicrobial resistance (AMR). CARD hosts ~4M pageviews each year and the web portal currently averages analyses of ~240 multidrug resistant pathogens per day and has been accessed by over 150 government or public health agencies, over 100 private sector companies, and over 1300 academic institutions. CARD has been cited over 8100 times amongst more than 150 different journals and currently averages ~60 citations per month.
Antimicrobial Resistance (AMR) Analytics. In 2014, Dr. McArthur initiated a new research program focused on the problem of antimicrobial resistance, building novel bioinformatics resources in preparation for the approaching transition to routine clinical and environmental DNA sequencing of important bacterial pathogens. Of particular impact has been the construction of the Comprehensive Antibiotic Resistance Database (latest publication: Alcock et al. 2023. Nucleic Acids Res. 51, D690-D699), a curated resource providing reference DNA and protein sequences, detection models and bioinformatics tools on the molecular basis of bacterial antimicrobial resistance (AMR). CARD hosts ~4M pageviews each year and the web portal currently averages analyses of ~240 multidrug resistant pathogens per day and has been accessed by over 150 government or public health agencies, over 100 private sector companies, and over 1300 academic institutions. CARD has been cited over 8100 times amongst more than 150 different journals and currently averages ~60 citations per month.
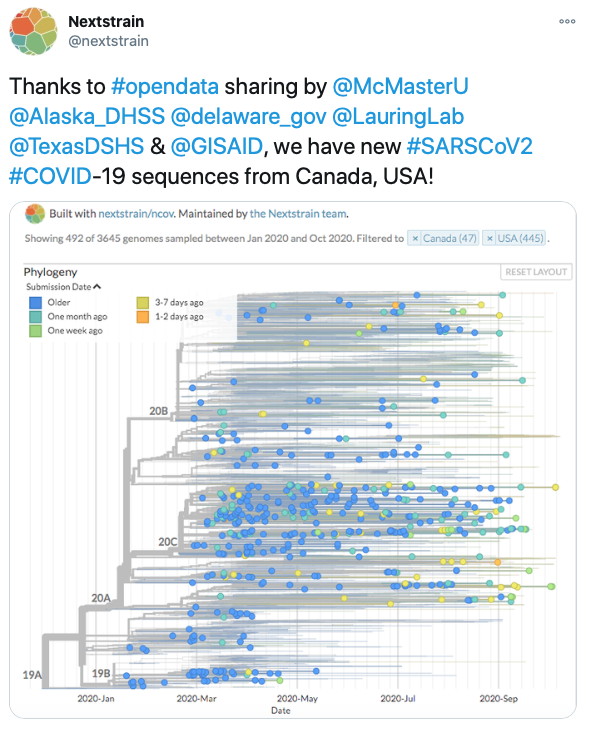 SARS-CoV-2 Molecular Epidemiology. The McArthur research team was a key member of our regional (Ontario) public health and research response to the COVID-19 pandemic. We co-led the Ontario SARS-CoV-2 genomic surveillance efforts for Ontario Genomics and the Public Health Agency of Ontario, helping develop standards and bioinformatics workflows for SARS-CoV-2 genome sequencing from clinical samples as well as being responsible for partnering with a dozen healthcare centres for sequencing of up to a third of Ontario positive cases in Year 1. On the research front, we provided key genome validation for isolation of the live SARS-CoV-2 virus (Banerjee et al. 2020. Emerging Infectious Diseases, 26, 2054-2063), developed machine learning approaches to SARS-CoV-2 molecular epidemiology (Mann et al. 2020. Lancet Digital Health, 2, e340–e341), and developed and released the SIGNAL Illumina workflow (Nasir et al. 2024. NAR Genome Bioinformatics 6, lqae176).
SARS-CoV-2 Molecular Epidemiology. The McArthur research team was a key member of our regional (Ontario) public health and research response to the COVID-19 pandemic. We co-led the Ontario SARS-CoV-2 genomic surveillance efforts for Ontario Genomics and the Public Health Agency of Ontario, helping develop standards and bioinformatics workflows for SARS-CoV-2 genome sequencing from clinical samples as well as being responsible for partnering with a dozen healthcare centres for sequencing of up to a third of Ontario positive cases in Year 1. On the research front, we provided key genome validation for isolation of the live SARS-CoV-2 virus (Banerjee et al. 2020. Emerging Infectious Diseases, 26, 2054-2063), developed machine learning approaches to SARS-CoV-2 molecular epidemiology (Mann et al. 2020. Lancet Digital Health, 2, e340–e341), and developed and released the SIGNAL Illumina workflow (Nasir et al. 2024. NAR Genome Bioinformatics 6, lqae176).