
Guitor et al. 2022. Microbiome 10(1):136. Background: Probiotic use in preterm infants can mitigate the impact of antibiotic exposure and reduce rates of certain illnesses;
Category: publications

big data clinical metadata epidemiology molecular epidemiology publications SARS-CoV-2 virus epidemiology
The Dataharmonizer: a Tool for Faster Data Harmonization, Validation, Aggregation, and Analysis of Pathogen Genomics Contextual Information
Gill et al. 2022. Preprints 10.20944/preprints202206.0335.v1 Pathogen genomics is a critical tool for public health surveillance, infection control, outbreak investigations, as well as research. In

Raphenya et al. Scientific Data volume 9, Article number: 341 (2022). Whole genome sequencing (WGS) is a key tool in identifying and characterising disease-associated bacteria

Rooney et al. 2022 Jun 1;e0002222. Short-read sequencing can provide detection of multiple genomic determinants of antimicrobial resistance from single bacterial genomes and metagenomic samples.

Kim et al. Clin Microbiol Rev. 2022 May 25;e0017921. Antimicrobial resistance (AMR) is a global health crisis that poses a great threat to modern medicine.

antibiotic resistance bioinformatics databases genomics molecular epidemiology publications software
Enabling genomic island prediction and comparison in multiple genomes to investigate bacterial evolution and outbreaks
Bertelli et al. Microb Genom. 2022 May;8(5). doi: 10.1099/mgen.0.000818. Outbreaks of virulent and/or drug-resistant bacteria have a significant impact on human health and major economic

Broadfield et al. Molecular Metabolism 2022. Apr 20;101498. Type 2 diabetes and obesity increase the risk of developing colorectal cancer (CRC). The first-line type 2

Sanderson et al. 2022. bioRxiv 10.1101/2022.04.11.487771. Enterococcus faecium is a ubiquitous opportunistic pathogen that is exhibiting increasing levels of antimicrobial resistance (AMR). Many of the

Kara K Tsang, Andrew Latchman, Nishma Singhal, Giuliana Federici, Sandra Russell, Denise Irwin, Robyn Stevens, Andrew G McArthur, & Sarah Khan BMC Med Educ. 2021

Wang, B., E.E. Tsakiridis, S. Zhang, A. Llanos, E.M. Desjardins, J.M. Yabut, A.E. Green, E.A. Day, B.K. Smith, J.S.V. Lally, J. Wu, A.R. Raphenya, K.A.
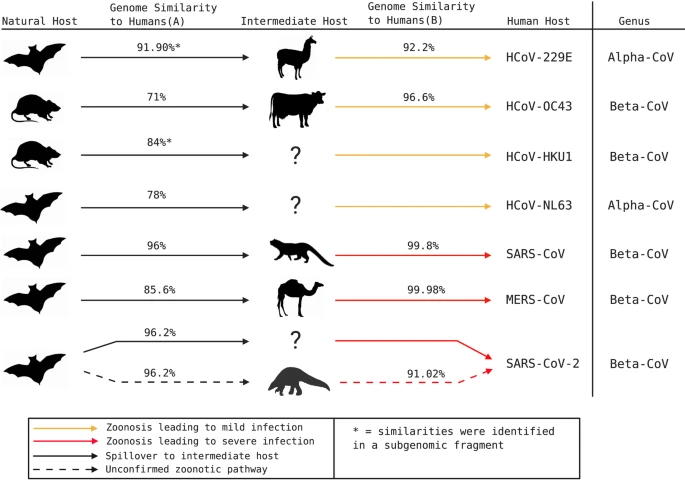
Jalen Singh, Pranav Pandit, Andrew G McArthur, Arinjay Banerjee, Karen Mossman Virology Journal. 2021 Aug 13;18(1):166. The emergence of a novel coronavirus, severe acute respiratory
Ashley M. Rooney, Amogelang R. Raphenya, Roberto G. Melano, Christine Seah, Noelle R. Yee, Derek R. MacFadden, Andrew G. McArthur, Pierre H.H. Schneeberger, Bryan Coburn.

Surface and air contamination with SARS-CoV-2 from hospitalized COVID-19 patients in Toronto, Canada
Kotwa et al. 2021. medRxiv https://doi.org/10.1101/2021.05.17.21257122 Background: The aim of this prospective cohort study was to determine the burden of SARS-CoV-2 in air and on

Pankov, K.V., A.G. McArthur, D.A. Gold, D.R. Nelson, J.V. Goldstone, J.Y. Wilson. Scientific Reports. 2021 May 10;11(1):9834. The cytochrome P450 (CYP) superfamily is a diverse

Sjaarda, C.P., J.L. Guthrie, S. Mubareka, J.T. Simpson, B. Hamelin, H. Wong, L. Mortimer, R. Slinger, A.G. McArthur, M. Desjardins, K. Douchant, Ontario’s COVID-19 Genomics

Banerjee, A., N. El-Sayes, P. Budylowski, D. Richard, H. Maan, J. Aguiar, K. Baid, M. D’Agostino, J. Ang, B. Tremblay, S. Afkhami, M. Karimzadeh, A.

Imagine a tiny molecular machine armed with a program to infect human cells, subvert their defenses and make copies of itself to infect neighboring cells
Kara K. Tsang, Finlay Maguire, Haley L. Zubyk, Sommer Chou, Arman Edalatmand, Gerard D. Wright, Robert G. Beiko, & Andrew G. McArthur Microbial Genomics, in

Hiu‐Ki R. Tran, Dirk W. Grebenc, Timothy A. Klein, John C. Whitney Molecular Microbiology, in press. Type VII secretion systems (T7SSs) are poorly understood protein

Charlie Tan, Fang-I Lu, Patryk Aftanas, Kara Tsang, Samira Mubareka, Adrienne Chan, & Robert Kozak IDCases. 2020. 23:e01034. We describe the case of a 33-year-old