
All online this year due to COVID-19, but some great presentations and virtual posters by our trainees and collaborators! Edalatmand, A. & A.G. McArthur. 2021.
Category: presentations

antibiotic resistance awards COVID19 lab members molecular epidemiology presentations SARS-CoV-2 virus epidemiology
WISE CREST, Ontario Biology Day, BBS Research Symposium 2021
McMaster Biochemistry & Biomedical Sciences Research Symposium Edalatmand. A. & A.G. McArthur. 2021. Ontology-driven method of contextualizing antimicrobial resistance determinants. Poster presentation at the McMaster

Aaron Petkau performed his Visual & Automated Disease Analytics Program internship (Univ. of Manitoba) with us during the summer of 2020, creating the CARD:Live graphic

antibiotic resistance bioinformatics COVID19 databases epidemiology genomics IIDR lab members molecular epidemiology presentations SARS-CoV-2 text mining virus epidemiology
IIDR Trainee Day 2020! Forging the Future of Infectious Disease Research
Nasir, J.A et al. 2020. SIGNALing the Need for Surveillance: A Comparison of Whole Genome Sequencing Methods for SARS-CoV-2. Edalatmand, A. & A.G. McArthur. 2020.
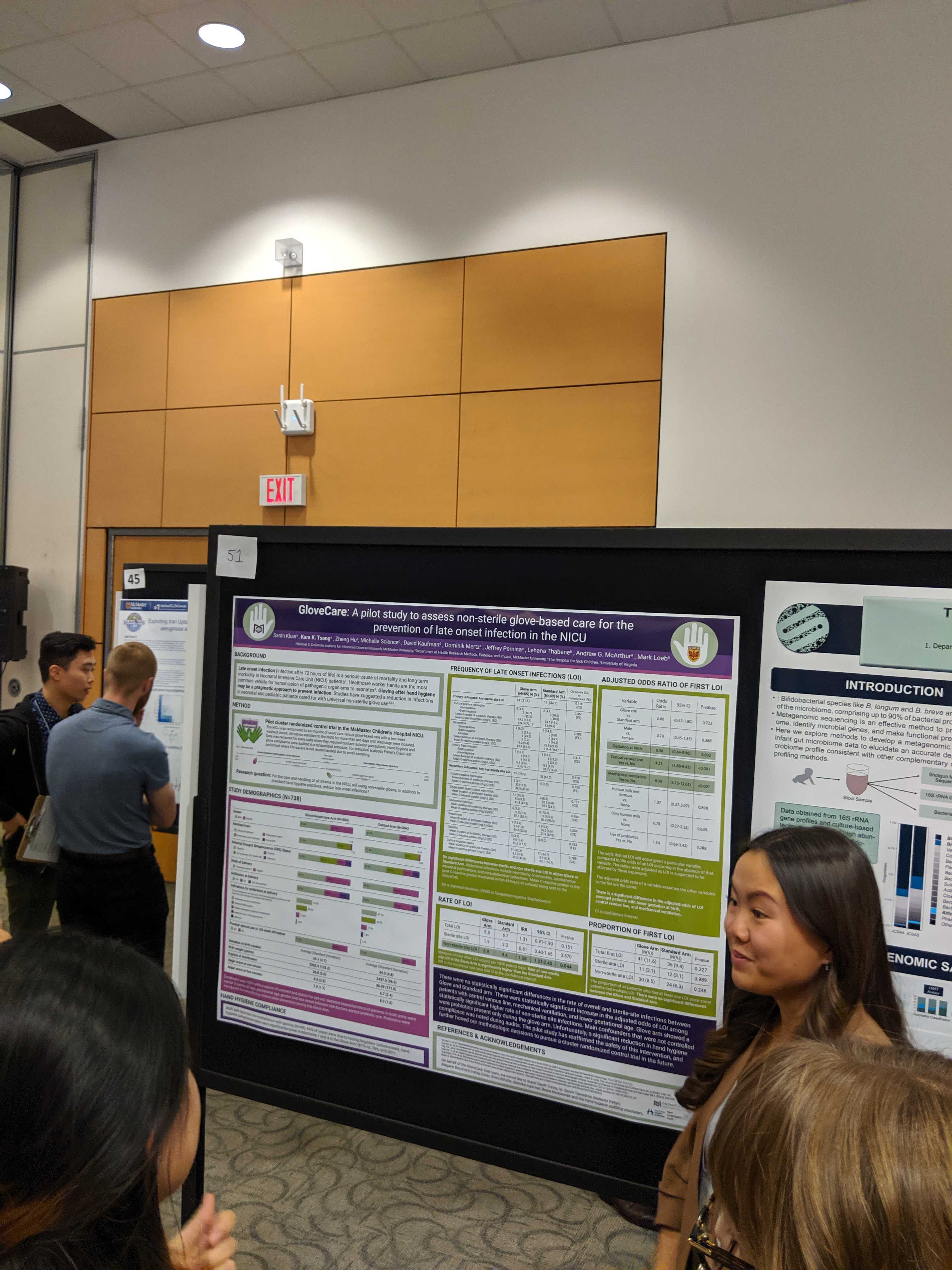
Edalatmand, A. & A.G. McArthur. 2019. Classifying and curating publications on antimicrobial resistance using machine learning. Poster presentation at the Michael G. DeGroote Institute for

Maguire, F., B. Alcock, A.R. Raphenya, B. Jia. E.J. Griffiths, T.C. Matthews, J. Adam, A. Petkau, G.L. Winsor, IRIDA Consortium, R.G. Beiko, F.S.L. Brinkman, W.W.L.
antibiotic resistance big data bioinformatics databases genomics lab members molecular epidemiology outreach presentations
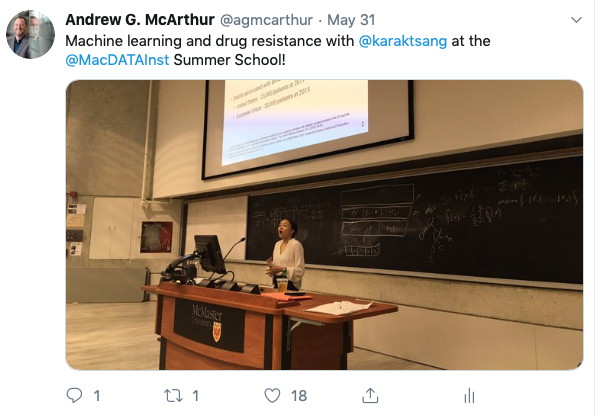
MacData Summer School 2019
Dr. McArthur and PhD student Kara Tsang taught together at the 2019 MacData Institute Summer School, with Dr. McArthur reviewing biocuration and bioinformatics for genomic

Speicher, D.J., K. Luinstra, J. Maciejewski, K.K. Tsang, A.G. McArthur, & M. Smieja. 2019. Clostridioides difficile strain divergence over time. Oral presentation at the Association of

Congratulations to Rachel Tran on winning a 2019 DBCAD Summer Fellowship! These competitive awards are designed to support students working in the labs of members
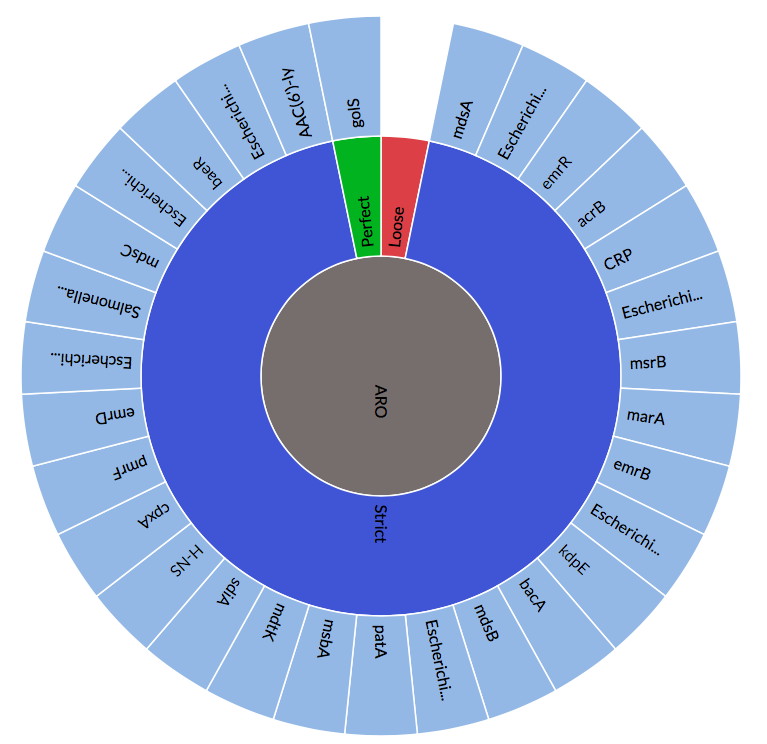
antibiotic resistance bioinformatics databases genomics IIDR molecular epidemiology ontologies presentations software teaching
State of the CARD 2019
A week of lectures, demos, and training for the Comprehensive Antibiotic Resistance Database During McMaster Spring Mid-Term Recess (February 18-24), the McArthur lab is pleased


A busy year for Dr. McArthur outside of academia… Integrated Health Biosystems Chair – Combatting Antimicrobial Resistance in the Age of Molecular Epidemiology. Invited presentation

Congratulations to #TeamVirulence for winning the 2018 McMaster Innovation Showcase People’s Choice Poster Award for their poster entitled, “Examining the relationship between virulence and antimicrobial resistance

Some invitations are more special than others. Dr. Peixoto da Cruz and I went to graduate school together in British Columbia (a long time ago!)

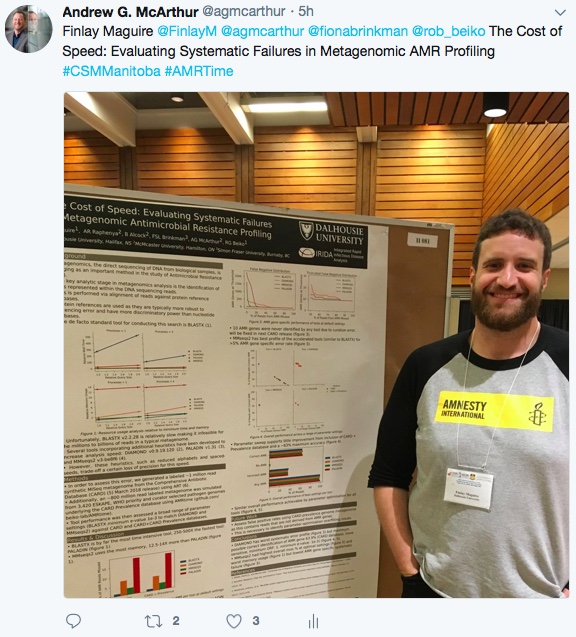
Maguire, F., B. Alcock, F.S. Brinkman, A.G. McArthur, & R.G. Beiko. 2018. AMRtime: Rapid Accurate Identification of Antimicrobial Resistance Determinants from Metagenomic Data. Oral presentation

Tsang, K.K., H. Zubyk, S. Chou, G.D. Wright, & A.G. McArthur. Decoding bad bags: Predicting antibiotic resistance phenotypes from genotype. Oral presentation at the Canadian Society of
Alcock, B., A.R. Raphenya, A.N. Sharma, K.K. Tsang, T.T.Y. Lau, A. Hernandez-Koutoucheva, & A.G. McArthur. 2018. Data and curation in the Comprehensive Antibiotic Resistance Database.

Dr. McArthur has been busy doing some Government outreach. In early 2018 he was a Panellist on Artificial Intelligence in Healthcare at Norwegian Health Ministry &

Florescu, A., B. Alcock, A. Raphenya, & A.G. McArthur. 2018. Incorporating phenotypic testing into ontological data sharing paradigms. Presentation at Ontario Biology Day, Waterloo, Ontario,

Tsang, K.K. & A.G. McArthur. 2017. Encoding the efflux pump phenomena. Oral presentation at the Second American Society for Microbiology Meeting on Rapid Applied Microbial