
Aaron Petkau performed his Visual & Automated Disease Analytics Program internship (Univ. of Manitoba) with us during the summer of 2020, creating the CARD:Live graphic
Category: databases

antibiotic resistance bioinformatics COVID19 databases epidemiology genomics IIDR lab members molecular epidemiology presentations SARS-CoV-2 text mining virus epidemiology
IIDR Trainee Day 2020! Forging the Future of Infectious Disease Research
Nasir, J.A et al. 2020. SIGNALing the Need for Surveillance: A Comparison of Whole Genome Sequencing Methods for SARS-CoV-2. Edalatmand, A. & A.G. McArthur. 2020.

antibiotic resistance bioinformatics databases genomics IIDR lab members molecular epidemiology ontologies
New CARD Team Member!
The McArthur lab welcomes new Curator-Developer William Huynh to the Comprehensive Antibiotic Resistance Database staff. Looking forward to pushing CARD forward with William’s help!

antibiotic resistance bioinformatics databases genomics IIDR lab members molecular epidemiology ontologies
Congratulations finishing thesis students!
COVID19 kept us from celebrating together, but we are very proud of the work you have accomplished! Rachel Tran | Biochem 4T15 – Exploring the

Chen CY, Clark CG, Langner S, Boyd DA, Bharat A, McCorrister SJ, McArthur AG, Graham MR, Westmacott GR, Van Domselaar G Proteomics Clin Appl. 2019

antibiotic resistance big data bioinformatics databases epidemiology genomics molecular epidemiology ontologies publications
CARD 2020: antibiotic resistome surveillance with the Comprehensive Antibiotic Resistance Database
Alcock BP, Raphenya AR, Lau TTY, Tsang KK, Bouchard M, Edalatmand A, Huynh W, Nguyen A-LV, Cheng AA, Liu S, Min SY, Miroshnichenko A, Tran
antibiotic resistance big data bioinformatics databases genomics lab members molecular epidemiology outreach presentations
MacData Summer School 2019
Dr. McArthur and PhD student Kara Tsang taught together at the 2019 MacData Institute Summer School, with Dr. McArthur reviewing biocuration and bioinformatics for genomic
The Comprehensive Antibiotic Resistance Database has been updated, http://card.mcmaster.ca CARD Curation: Expanded MCR, OXA & IMP beta-lactamase, and macrolide phosphotransferase (MPH) sequence curation. Updated nomenclature
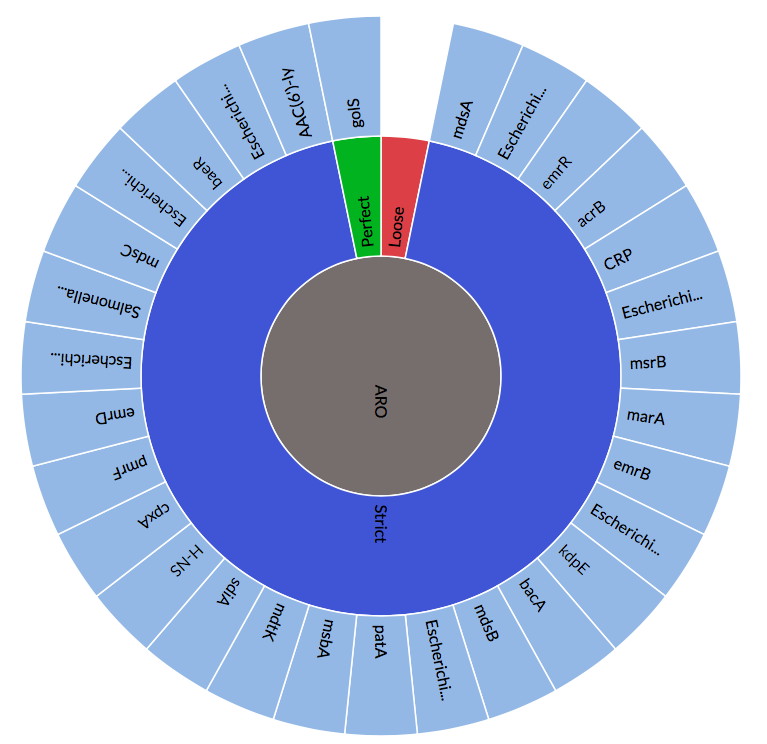
antibiotic resistance bioinformatics databases genomics IIDR molecular epidemiology ontologies presentations software teaching
State of the CARD 2019
A week of lectures, demos, and training for the Comprehensive Antibiotic Resistance Database During McMaster Spring Mid-Term Recess (February 18-24), the McArthur lab is pleased

Antimicrobial Resistance: Emergence, Transmission, and Ecology (ARETE). R. Beiko (PI; Dalhousie University), F. Brinkman (co-PI, Simon Fraser University), A.G. McArthur (co-Applicant) + 4 additional co-Applicants.

Congratulations to #TeamVirulence for winning the 2018 McMaster Innovation Showcase People’s Choice Poster Award for their poster entitled, “Examining the relationship between virulence and antimicrobial resistance

Some invitations are more special than others. Dr. Peixoto da Cruz and I went to graduate school together in British Columbia (a long time ago!)

Welcome #TeamVirulence, left to right: Rachel Tran (Biochem 3R06), Sally Min (BiomedDC 4A15), Anatoly Miroshnichencko (BiomedDC 4A15), and Rafik El Werfalli (BiomedDC 4A15), who are

Tsang, K.K., H. Zubyk, S. Chou, G.D. Wright, & A.G. McArthur. Decoding bad bags: Predicting antibiotic resistance phenotypes from genotype. Oral presentation at the Canadian Society of
Alcock, B., A.R. Raphenya, A.N. Sharma, K.K. Tsang, T.T.Y. Lau, A. Hernandez-Koutoucheva, & A.G. McArthur. 2018. Data and curation in the Comprehensive Antibiotic Resistance Database.

Kara Tsang passed her graduate transfer exam today, officially moving from the McMaster Biochemistry & Biomedical Sciences Masters program to the Ph.D. program. Kara’s work
antibiotic resistance big data bioinformatics databases epidemiology genomics molecular epidemiology news software
Recent Updates to the Comprehensive Antibiotic Resistance Database
The Comprehensive Antibiotic Resistance Database has been updated, http://card.mcmaster.ca CARD Curation: Addition of HERA, TRU, & ACI beta-lactamases, sul4, and new quinolone efflux pumps. Antibiotic
antibiotic resistance big data bioinformatics databases epidemiology genomics molecular epidemiology news software
Major Update of the Comprehensive Antibiotic Resistance Database
The Comprehensive Antibiotic Resistance Database has been updated, http://card.mcmaster.ca This February 2018 release is our largest to date and includes new data types, a new

Tsang, K.K. & A.G. McArthur. 2017. Encoding the efflux pump phenomena. Oral presentation at the Second American Society for Microbiology Meeting on Rapid Applied Microbial

Today we say farewell to Arjun Sharma & Suman Virdee. Arjun joined the lab as a second year volunteer, staying to perform a Biochem 3R06