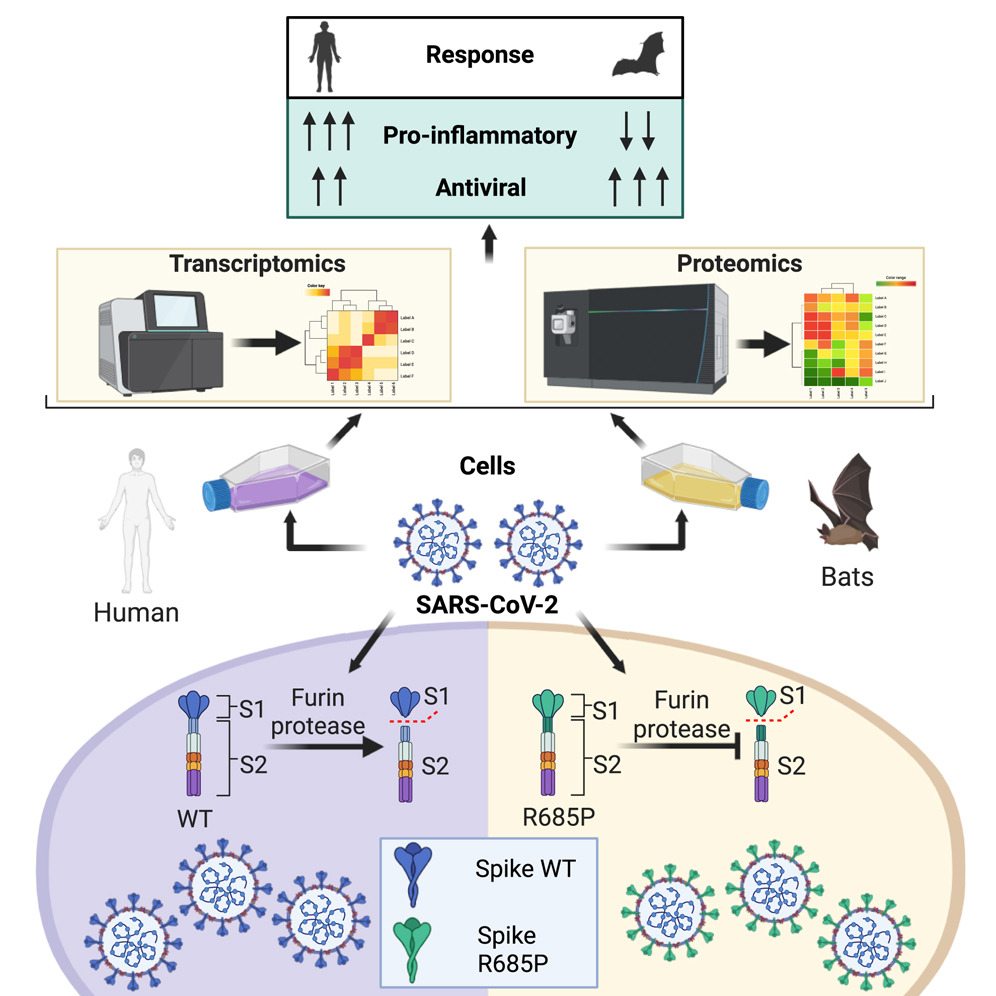
Baid et al. 2015. Cell Reports 44, 115929. SARS-CoV-2 has caused the largest known coronavirus pandemic and is believed to have emerged from insectivorous bats.
Category: COVID19

It is with great pleasure that we announce that Dr. Jalees Nasir has been awarded the 2025 McMaster Health Sciences Graduate Student Association (HSGSA) Impact

bioinformatics COVID19 genomics lab members molecular epidemiology SARS-CoV-2 software virus epidemiology
Congratulations Dr. Jalees Nasir!
Designing Molecular Fishhooks for Virus Survellance Platforms By JALEES A. NASIR, B.Sc A Thesis Submitted to the School of Graduate Studies in partial fulfilment of

This week we say farewell to Dr. Sheridan Baker, who joined us in early 2021 as lead molecular biologist for our Ontario Genomics Coalition (ONCoV)

Nirmalarajah et al. BMC Infect Dis. 2025 Jan 28;25(1):132. Background: Drivers of COVID-19 severity are multifactorial and include multidimensional and potentially interacting factors encompassing viral

Nasir et al. NAR Genomics & Bioinformatics. 2024 Dec 18;6(4):lqae176. The incorporation of sequencing technologies in frontline and public health healthcare settings was vital in

Gill et al. 2024. The Canadian VirusSeq Data Portal & Duotang: open resources for SARS-CoV-2 viral sequences and genomic epidemiology. Microb Genom. 2024 Oct;10(10):001293. The
clinical metadata COVID19 genomics molecular epidemiology publications SARS-CoV-2 virus epidemiology
Chronic COVID-19 infection in an immunosuppressed patient shows changes in lineage over time: a case report
Sheridan J C Baker , Landry E Nfonsam , Daniela Leto , Candy Rutherford , Marek Smieja , & Andrew G McArthur Virol J. 2024

Baker, S.J.C., J. Maciejewski, M.-T. Usuanlele, J. Gilchrist, D.R. Sharma, D. Bulir, M. Smieja, M. Loeb, M.G. Surette, A.G. McArthur, & D. Mertz. 2023. Investigating

Jacob et al. Emerg Infect Dis. 2023 Jun 12;29(7). Isolating and characterizing emerging SARS-CoV-2 variants is key to understanding virus pathogenesis. In this study, we

The SARS-CoV-2 Illumina GeNome Assembly Line (SIGNAL) has been updated to version 1.6.2, with an important hotfix to address software version incompatibility for rules using

Tsang et al. Microbiol Spectr. 2023 Apr 24;e0190022. Genomic epidemiology can facilitate an understanding of evolutionary history and transmission dynamics of a severe acute respiratory
awards bioinformatics COVID19 epidemiology genomics lab members molecular epidemiology SARS-CoV-2 software virus epidemiology
Congratulations Ph.D. student Jalees Nasir!
Congratulations Jalees Nasir on being awarded the Dr. Jordon Page Harshman Bursary! The Dr. Jordan Page Harsham Bursary was established in 2011 by the Harshman

Congratulations Dr. Sheridan Baker on being awarded a 2-year Mitacs Elevate Postdoctoral Fellowship, which will allow him to train jointly in clinical epidemiology, molecular epidemiology,
Emma J. Griffiths et al. Gigascience. 2022 Feb 16;11:giac003. Background: The Public Health Alliance for Genomic Epidemiology (PHA4GE) (https://pha4ge.org) is a global coalition that is

Kara K Tsang, Andrew Latchman, Nishma Singhal, Giuliana Federici, Sandra Russell, Denise Irwin, Robyn Stevens, Andrew G McArthur, & Sarah Khan BMC Med Educ. 2021

awards COVID19 epidemiology genomics IIDR lab members molecular epidemiology SARS-CoV-2 virus epidemiology
Emily Panousis wins the Michael Kamin Hart Memorial Undergraduate Scholarship for her work on SARS-CoV-2 surveillance!
More information about IIDR Trainee Day and Michael Kamin Hart here.
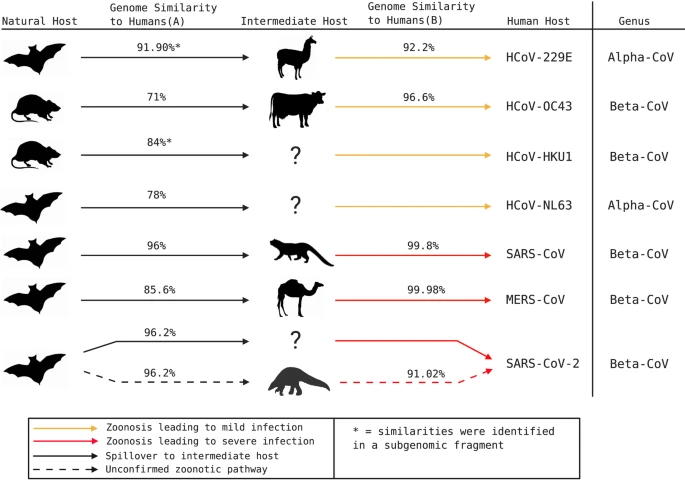
Jalen Singh, Pranav Pandit, Andrew G McArthur, Arinjay Banerjee, Karen Mossman Virology Journal. 2021 Aug 13;18(1):166. The emergence of a novel coronavirus, severe acute respiratory

Surface and air contamination with SARS-CoV-2 from hospitalized COVID-19 patients in Toronto, Canada
Kotwa et al. 2021. medRxiv https://doi.org/10.1101/2021.05.17.21257122 Background: The aim of this prospective cohort study was to determine the burden of SARS-CoV-2 in air and on

Sjaarda, C.P., J.L. Guthrie, S. Mubareka, J.T. Simpson, B. Hamelin, H. Wong, L. Mortimer, R. Slinger, A.G. McArthur, M. Desjardins, K. Douchant, Ontario’s COVID-19 Genomics