
Mukiri, K.M., B.P. Alcock, & A.G. McArthur. 2024. Increasing the predictive accuracy of the Resistance Gene Identifier by abandoning sole reliance on bitscore. Ta, T.E.,

Congratulations to Colin Bruce for winning First Place PhD poster at the Biochemistry & Biomedical Sciences Research Symposium!

Allison K Guitor, Anna Katyukhina, Margaret Mokomane, Kwana Lechiile, David M Goldfarb, Gerard D Wright, Andrew G McArthur, & Jeffrey M Pernica J Infect Dis.
clinical metadata COVID19 genomics molecular epidemiology publications SARS-CoV-2 virus epidemiology
Chronic COVID-19 infection in an immunosuppressed patient shows changes in lineage over time: a case report
Sheridan J C Baker , Landry E Nfonsam , Daniela Leto , Candy Rutherford , Marek Smieja , & Andrew G McArthur Virol J. 2024

Day, E.A., L.K. Townsend, S. Rehal, B. Batchuluun, D. Wang, M.R. Morrow, R. Lu, L. Lundenberg, J.H. Lu, E.M. Desjardins, A.R. Raphenya, A.G. McArthur, M.D.

Keaton W Smith, Brian P Alcock, Shawn French, Maya A Farha, Amogelang R Raphenya, Eric D Brown, & Andrew G McArthur. Microbiol Spectrum. 2023 Nov

antibiotic resistance big data bioinformatics databases epidemiology genomics IIDR lab members molecular epidemiology ontologies presentations software text mining
IIDR Trainee Day 2023
COLIN BRUCE – Investigating Fibre Degradation in the Infant Gut Microbiota ; DIRK HACKENBERGER – Was World War 2 Foundational to the Antimicrobial Resistance Crisis?
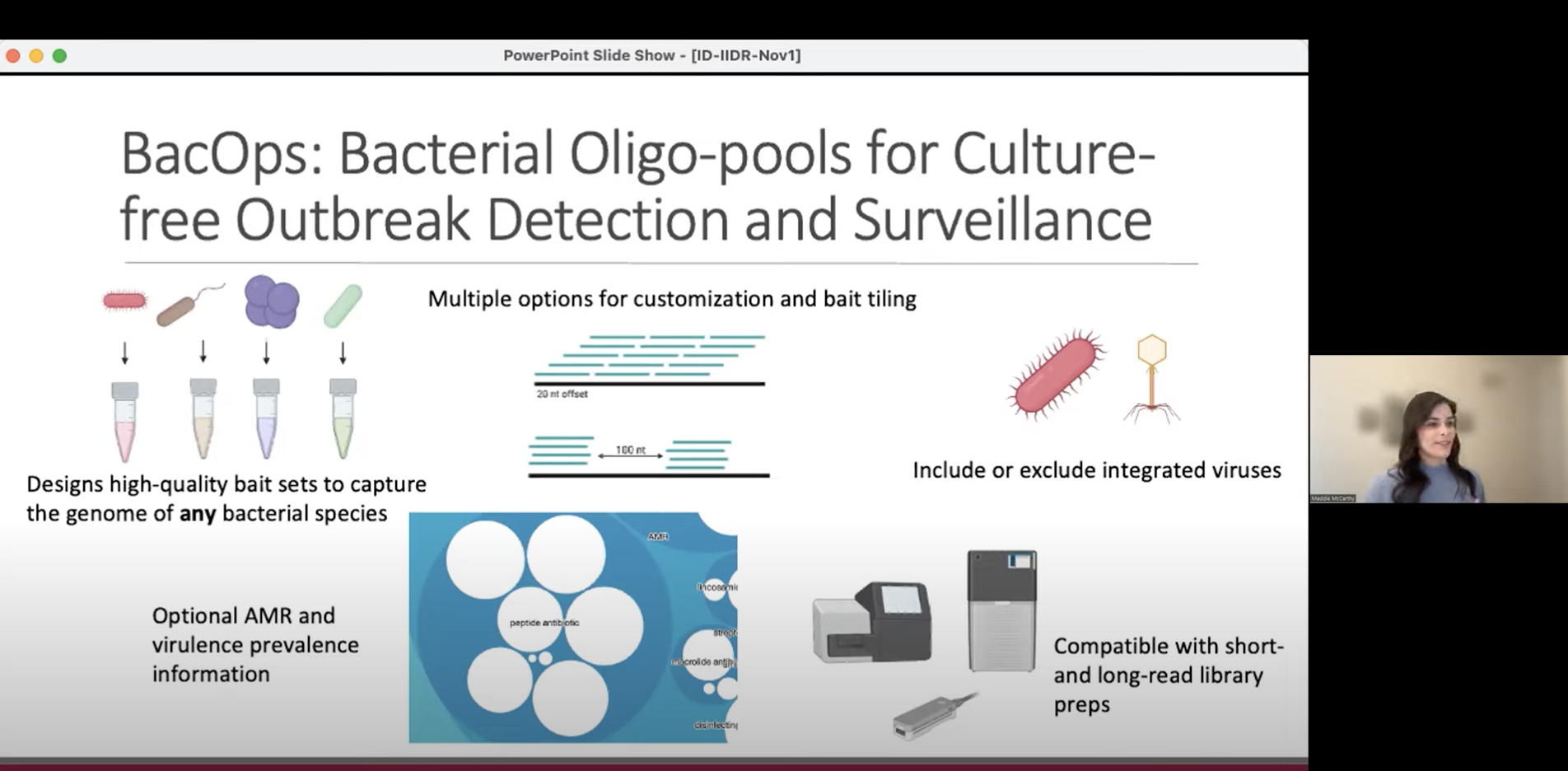
ID-IIDR Rounds – Clostridioides (Clostridium) difficile: new insights from the clinic and laboratory
This one-hour webinar features talks from Marek Smieja (Professor, Pathology & Molecular Medicine), Sheridan Baker (Postdoctoral Fellow, The McArthur Lab and Smieja Lab), and Maddie

2023-2024 Thesis & Inquiry Course Students Kriti Goel (3rd year Biochemistry) – BIOCHEM 3R06 Tiffany Ta (4th year Biomedical Discovery & Commercialization) – BIOMEDDC 4A15 Kristine

The McArthur lab welcomes Brody Duncan, M.D. (Hamilton Health Sciences) as he starts his M.Sc. studies with us, investigating standards and methods for clinical reporting