
Komorowski et al. 2025. Journal of Infectious Diseases, in press. Background: Serratia marcescens is an opportunistic AmpC β-lactamase-producing Enterobacterales associated with intensive care unit outbreaks,
Category: publications

Baker et al. 2025. Journal of the Association of Medical Microbiology and Infectious Disease Canada, in press. Background: Clostridioides difficile is a bacillus that can
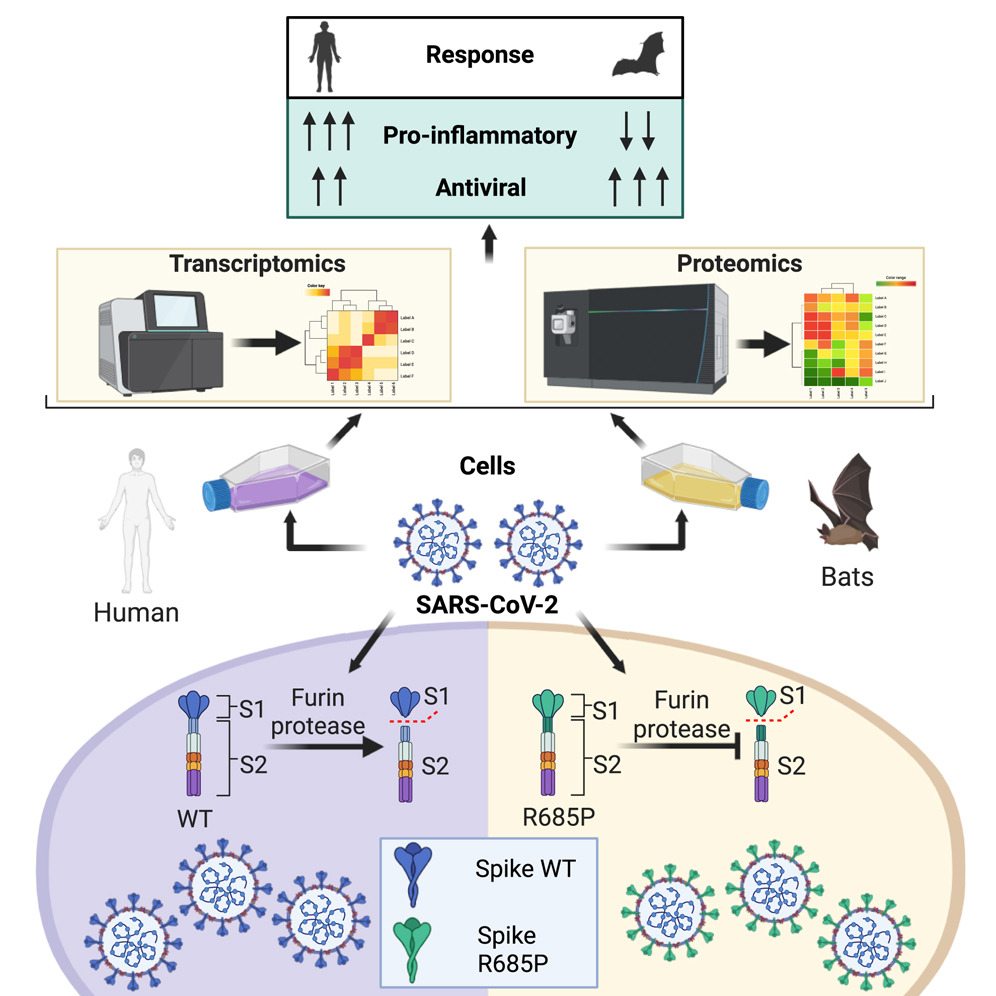
Baid et al. 2015. Cell Reports 44, 115929. SARS-CoV-2 has caused the largest known coronavirus pandemic and is believed to have emerged from insectivorous bats.

Baker et al. IDCases. 2025 May 28:40:e02274. Background: Differentiating severe systemic inflammatory syndromes from sepsis can be challenging. The diagnostic process may be further complicated

Lu et al. Genome Med. 2025 May 6;17(1):46. Background: Antimicrobial resistant (AMR) pathogens represent urgent threats to human health, and their surveillance is of paramount

Hackenberger et al. Appl Environ Microbiol. 2025 Feb 28:e0187624. Better interrogation of antimicrobial resistance requires new approaches to detect the associated genes in metagenomic samples.

This week we say farewell to Dr. Sheridan Baker, who joined us in early 2021 as lead molecular biologist for our Ontario Genomics Coalition (ONCoV)

Nirmalarajah et al. BMC Infect Dis. 2025 Jan 28;25(1):132. Background: Drivers of COVID-19 severity are multifactorial and include multidimensional and potentially interacting factors encompassing viral

Nasir et al. NAR Genomics & Bioinformatics. 2024 Dec 18;6(4):lqae176. The incorporation of sequencing technologies in frontline and public health healthcare settings was vital in
Tsang et al. Microb Genom. 2024 Oct;10(10). Interpreting the phenotypes of bla SHV alleles in Klebsiella pneumoniae genomes is complex. Whilst all strains are expected

Gill et al. 2024. The Canadian VirusSeq Data Portal & Duotang: open resources for SARS-CoV-2 viral sequences and genomic epidemiology. Microb Genom. 2024 Oct;10(10):001293. The

Gill, E.E., B. Jia, C.L. Murall, R. Poujol, M.Z. Anwar, N.S. John, J. Richardsson, A. Hobb, A.S. Olabode, A. Lepsa, A.T. Duggan, A.D. Tyler, A.

Allison K Guitor, Anna Katyukhina, Margaret Mokomane, Kwana Lechiile, David M Goldfarb, Gerard D Wright, Andrew G McArthur, & Jeffrey M Pernica J Infect Dis.
clinical metadata COVID19 genomics molecular epidemiology publications SARS-CoV-2 virus epidemiology
Chronic COVID-19 infection in an immunosuppressed patient shows changes in lineage over time: a case report
Sheridan J C Baker , Landry E Nfonsam , Daniela Leto , Candy Rutherford , Marek Smieja , & Andrew G McArthur Virol J. 2024

Day, E.A., L.K. Townsend, S. Rehal, B. Batchuluun, D. Wang, M.R. Morrow, R. Lu, L. Lundenberg, J.H. Lu, E.M. Desjardins, A.R. Raphenya, A.G. McArthur, M.D.

Keaton W Smith, Brian P Alcock, Shawn French, Maya A Farha, Amogelang R Raphenya, Eric D Brown, & Andrew G McArthur. Microbiol Spectrum. 2023 Nov

Jacob et al. Emerg Infect Dis. 2023 Jun 12;29(7). Isolating and characterizing emerging SARS-CoV-2 variants is key to understanding virus pathogenesis. In this study, we

Tsang et al. Microbiol Spectr. 2023 Apr 24;e0190022. Genomic epidemiology can facilitate an understanding of evolutionary history and transmission dynamics of a severe acute respiratory

Arman Edalatmand & Andrew G McArthur. 2023. Database, doi: 10.1093/database/baad023. Scientific literature is published at a rate that makes manual data extraction a highly time-consuming

Alcock et al. 2023. Nucleic Acids Res Oct 20, 2022:gkac920. (online ahead of print) The Comprehensive Antibiotic Resistance Database (CARD; card.mcmaster.ca) combines the Antibiotic Resistance