
Mateusz Wlodarski wins the bioMerieux prize for best poster presentation! Arnold, A., A.R. Raphenya, A.G. McArthur, & J.M. Stokes. 2024. The ESKAPE model: AI-guided antibiotic
Category: epidemiology

antibiotic resistance big data bioinformatics databases epidemiology genomics IIDR lab members molecular epidemiology ontologies presentations software text mining
IIDR Trainee Day 2023
COLIN BRUCE – Investigating Fibre Degradation in the Infant Gut Microbiota ; DIRK HACKENBERGER – Was World War 2 Foundational to the Antimicrobial Resistance Crisis?

awards bioinformatics COVID19 epidemiology genomics lab members molecular epidemiology SARS-CoV-2 software virus epidemiology
Congratulations Ph.D. student Jalees Nasir!
Congratulations Jalees Nasir on being awarded the Dr. Jordon Page Harshman Bursary! The Dr. Jordan Page Harsham Bursary was established in 2011 by the Harshman

antibiotic resistance awards bioinformatics Co-Op positions databases epidemiology genomics IIDR lab members ontologies presentations
IIDR Trainee Day 2022!
KARYN MUKIRI – Predicting the total resistome KEATON SMITH – Advancements in curation of the Comprehensive Antibiotic Resistance Database JALEES NASIR – Viral fishing expedition:


big data clinical metadata epidemiology molecular epidemiology publications SARS-CoV-2 virus epidemiology
The Dataharmonizer: a Tool for Faster Data Harmonization, Validation, Aggregation, and Analysis of Pathogen Genomics Contextual Information
Gill et al. 2022. Preprints 10.20944/preprints202206.0335.v1 Pathogen genomics is a critical tool for public health surveillance, infection control, outbreak investigations, as well as research. In

awards COVID19 epidemiology genomics IIDR lab members molecular epidemiology SARS-CoV-2 virus epidemiology
Emily Panousis wins the Michael Kamin Hart Memorial Undergraduate Scholarship for her work on SARS-CoV-2 surveillance!
More information about IIDR Trainee Day and Michael Kamin Hart here.

awards bioinformatics epidemiology genomics IIDR lab members news SARS-CoV-2 software virus epidemiology
Jalees Nasir awarded 2021 Fred & Helen Knight Enrichment Award!
Congratulations to PhD student Jalees Nasir, who has been awarded the prestigious 2021 Fred & Helen Knight Enrichment Award, which recognizes exceptional promise in early
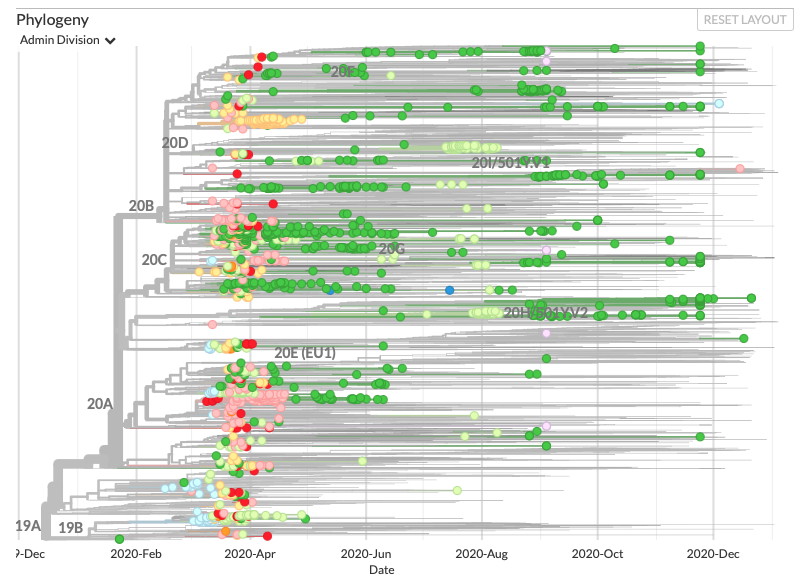
In collaboration with our colleague Dr. Finlay Maguire (Dalhousie University) and as part of our ongoing work for Ontario’s COVID-19 Genomics Rapid Response Coalition and the Canadian

antibiotic resistance bioinformatics COVID19 databases epidemiology genomics IIDR lab members molecular epidemiology presentations SARS-CoV-2 text mining virus epidemiology
IIDR Trainee Day 2020! Forging the Future of Infectious Disease Research
Nasir, J.A et al. 2020. SIGNALing the Need for Surveillance: A Comparison of Whole Genome Sequencing Methods for SARS-CoV-2. Edalatmand, A. & A.G. McArthur. 2020.
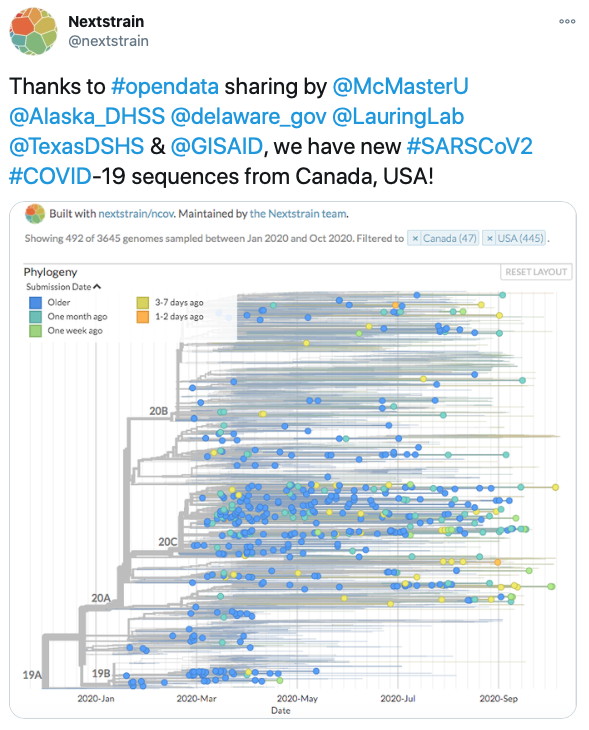

bioinformatics COVID19 epidemiology genomics molecular epidemiology publications SARS-CoV-2 software virus epidemiology
A Comparison of Whole Genome Sequencing of SARS-CoV-2 Using Amplicon-Based Sequencing, Random Hexamers, and Bait Capture
Jalees A. Nasir, Robert A. Kozak, Patryk Aftanas, Amogelang R. Raphenya, Kendrick M. Smith, Finlay Maguire, Hassaan Maan, Muhannad Alruwaili, Arinjay Banerjee, Hamza Mbareche, Brian

Emma J. Griffiths, Ruth E. Timme, Andrew J. Page, Nabil-Fareed Alikhan, Dan Fornika, Finlay Maguire, Catarina Inês Mendes, Simon H. Tausch, Allison Black, Thomas R.

The McArthur lab welcomes Ahmed Draia for an internship placement as part of McMaster’s Masters of Biomedical Discovery & Commercialization (MBDC) program. Reflective of his
antibiotic resistance COVID19 epidemiology genomics lab members molecular epidemiology news SARS-CoV-2 virus epidemiology
McArthur Lab & CARD during SARS-CoV-2
Quick update on the status of the McArthur Lab and the Comprehensive Antibiotic Resistance Database. While our home institution McMaster University is closed to on-site

antibiotic resistance big data bioinformatics databases epidemiology genomics molecular epidemiology ontologies publications
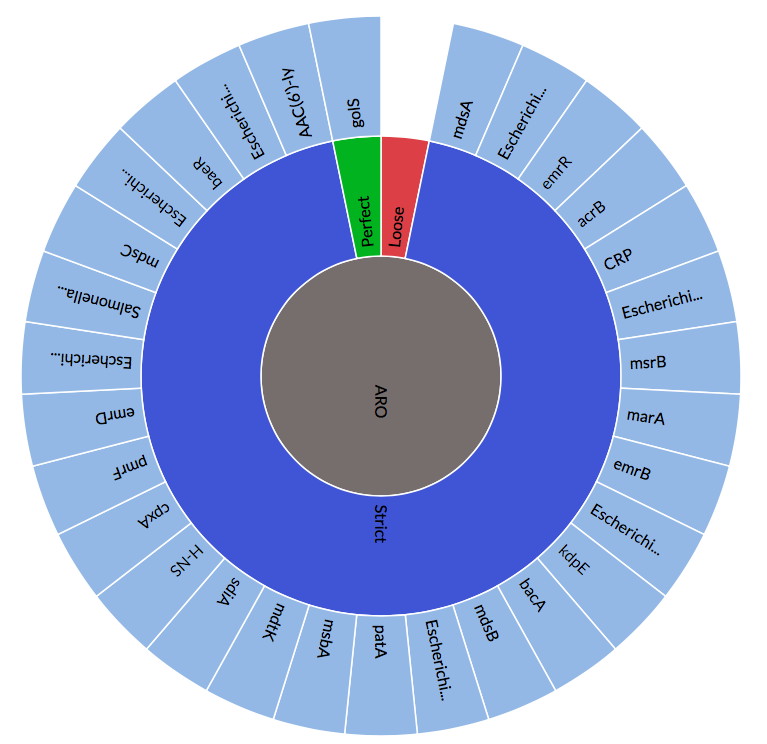
CARD 2020: antibiotic resistome surveillance with the Comprehensive Antibiotic Resistance Database
Alcock BP, Raphenya AR, Lau TTY, Tsang KK, Bouchard M, Edalatmand A, Huynh W, Nguyen A-LV, Cheng AA, Liu S, Min SY, Miroshnichenko A, Tran

Speicher, D.J., K. Luinstra, J. Maciejewski, K.K. Tsang, A.G. McArthur, & M. Smieja. 2019. Clostridioides difficile strain divergence over time. Oral presentation at the Association of
antibiotic resistance big data bioinformatics databases epidemiology genomics molecular epidemiology news software
Recent Updates to the Comprehensive Antibiotic Resistance Database
The Comprehensive Antibiotic Resistance Database has been updated, http://card.mcmaster.ca CARD Curation: Addition of HERA, TRU, & ACI beta-lactamases, sul4, and new quinolone efflux pumps. Antibiotic
The Comprehensive Antibiotic Resistance Database: Expanded tools for molecular surveillance of antimicrobial resistance (AMR) in the environment, agriculture, and clinic. A.G. McArthur (PI), G. Van

The McArthur lab and the Comprehensive Antibiotic Resistance Database are proud to join the Canadian Anti-Infective Innovation Network, International Genomic Epidemiology Application Ontology Consortium, and Integrated Rapid