
Characterizing the Influence of Dietary Fibre Consumption on the Composition and Function of Fibre Fermenting Bacteria in the Infant Gut Microbiome By COLIN Y. BRUCE,
Category: genomics

Komorowski et al. 2025. Journal of Infectious Diseases, in press. Background: Serratia marcescens is an opportunistic AmpC β-lactamase-producing Enterobacterales associated with intensive care unit outbreaks,

Baker et al. 2025. Journal of the Association of Medical Microbiology and Infectious Disease Canada, in press. Background: Clostridioides difficile is a bacillus that can

antibiotic resistance bioinformatics genomics lab members molecular epidemiology presentations software text mining
33rd Conference on Intelligent Systems for Molecular Biology
Mukiri, K., B. Alcock, A. Raphenya, & A.G. McArthur. 2025. Mind the gap: predicting the total bacterial resistome in the fight against antimicrobial resistance. Poster

Baker et al. IDCases. 2025 May 28:40:e02274. Background: Differentiating severe systemic inflammatory syndromes from sepsis can be challenging. The diagnostic process may be further complicated

It is with great pleasure that we announce that Dr. Jalees Nasir has been awarded the 2025 McMaster Health Sciences Graduate Student Association (HSGSA) Impact

Lu et al. Genome Med. 2025 May 6;17(1):46. Background: Antimicrobial resistant (AMR) pathogens represent urgent threats to human health, and their surveillance is of paramount

Antibiotics and Resistance: Past, Present, and Future By DIRK L HACKENBERGER, B.Sc. A thesis submitted to the school of graduate studies in partial fulfillment of

bioinformatics COVID19 genomics lab members molecular epidemiology SARS-CoV-2 software virus epidemiology
Congratulations Dr. Jalees Nasir!
Designing Molecular Fishhooks for Virus Survellance Platforms By JALEES A. NASIR, B.Sc A Thesis Submitted to the School of Graduate Studies in partial fulfilment of


Wlodarski, M.A., T.T.Y. Lau, B.P. Alcock, A.R. Raphenya, F. Maguire, R.G. Beiko, T.E. Ta, & A.G. McArthur. 2025. Unmasking antibiotic resistance genes and their pathogen

Hackenberger et al. Appl Environ Microbiol. 2025 Feb 28:e0187624. Better interrogation of antimicrobial resistance requires new approaches to detect the associated genes in metagenomic samples.

Two alumni of the McMaster Biochemistry graduate program have joined the McArthur Lab! Dr. Emily Bordeleau got her PhD from McMaster in 2022 under the

This week we say farewell to Dr. Sheridan Baker, who joined us in early 2021 as lead molecular biologist for our Ontario Genomics Coalition (ONCoV)

Nirmalarajah et al. BMC Infect Dis. 2025 Jan 28;25(1):132. Background: Drivers of COVID-19 severity are multifactorial and include multidimensional and potentially interacting factors encompassing viral

Nasir et al. NAR Genomics & Bioinformatics. 2024 Dec 18;6(4):lqae176. The incorporation of sequencing technologies in frontline and public health healthcare settings was vital in

Mateusz Wlodarski wins the bioMerieux prize for best poster presentation! Arnold, A., A.R. Raphenya, A.G. McArthur, & J.M. Stokes. 2024. The ESKAPE model: AI-guided antibiotic
antibiotic resistance bioinformatics databases genomics IIDR lab members molecular epidemiology ontologies presentations software volunteers
IIDR Trainee Day 2024!
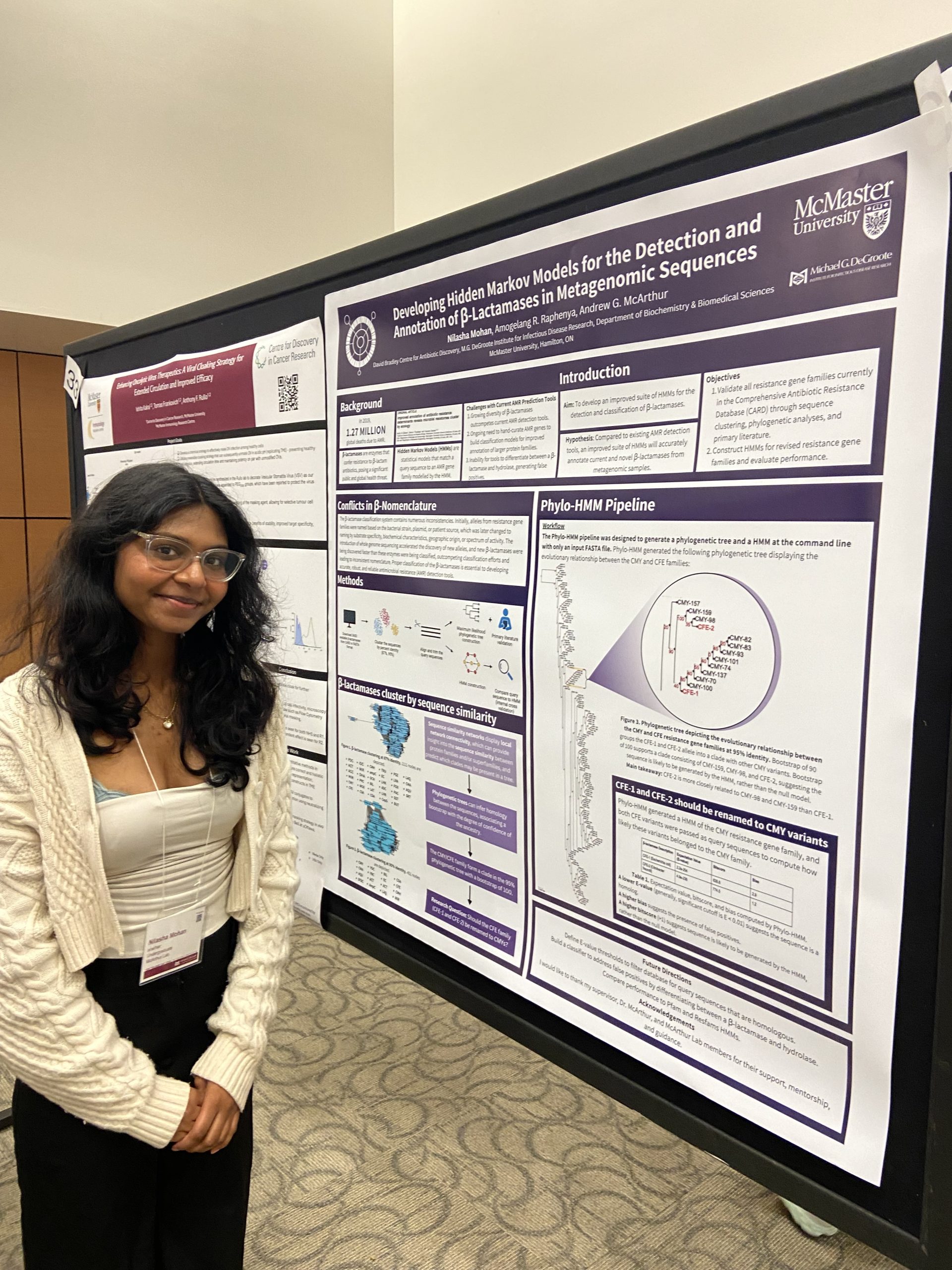
A great day of posters and presentations by the trainees of the Michael G. DeGroote Institute of Infectious Disease Research! Nilasha Mohan, Undergraduate student. Developing
Tsang et al. Microb Genom. 2024 Oct;10(10). Interpreting the phenotypes of bla SHV alleles in Klebsiella pneumoniae genomes is complex. Whilst all strains are expected

Gill et al. 2024. The Canadian VirusSeq Data Portal & Duotang: open resources for SARS-CoV-2 viral sequences and genomic epidemiology. Microb Genom. 2024 Oct;10(10):001293. The