
Gill et al. 2024. The Canadian VirusSeq Data Portal & Duotang: open resources for SARS-CoV-2 viral sequences and genomic epidemiology. Microb Genom. 2024 Oct;10(10):001293. The
Category: genomics
Wlodarski, M.A., T.T.Y. Lau, A.R. Raphenya, B.P. Alcock, & A.G. McArthur. 2024. Accurate pathogen-of-origin classification of antibiotic resistance genes with CARD k-mers. Presentation at the

This one-hour webinar, entitled “DNA sequencing in infectious disease surveillance, diagnosis, and management,” features talks from Andrew McArthur (Professor, Biochemistry and Biomedical Sciences) and Rubayet

CSM Mukiri, K.M., B.P. Alcock, & A.G. McArthur. 2024. Increasing the predictive accuracy of the Resistance Gene Identifier by abandoning sole reliance on bitscore.

Gill, E.E., B. Jia, C.L. Murall, R. Poujol, M.Z. Anwar, N.S. John, J. Richardsson, A. Hobb, A.S. Olabode, A. Lepsa, A.T. Duggan, A.D. Tyler, A.

Alcock, B.P., E.A. Bordeleau, & A.G McArthur. 2024. Improving aminoglycoside resistance surveillance and stewardship efforts through nomenclature harmonization with the Comprehensive Antibiotic Resistance Database. Presentation

antibiotic resistance big data bioinformatics databases genomics molecular epidemiology presentations
Antimicrobial Resistance – Genomes, Big Data and Emerging Technologies – Wellcome Trust, UK
Mukiri, K.M., B.P. Alcock, & A.G. McArthur. 2024. Increasing the predictive accuracy of the Resistance Gene Identifier by abandoning sole reliance on bitscore. Ta, T.E.,

Allison K Guitor, Anna Katyukhina, Margaret Mokomane, Kwana Lechiile, David M Goldfarb, Gerard D Wright, Andrew G McArthur, & Jeffrey M Pernica J Infect Dis.
clinical metadata COVID19 genomics molecular epidemiology publications SARS-CoV-2 virus epidemiology
Chronic COVID-19 infection in an immunosuppressed patient shows changes in lineage over time: a case report
Sheridan J C Baker , Landry E Nfonsam , Daniela Leto , Candy Rutherford , Marek Smieja , & Andrew G McArthur Virol J. 2024

antibiotic resistance big data bioinformatics databases epidemiology genomics IIDR lab members molecular epidemiology ontologies presentations software text mining
IIDR Trainee Day 2023
COLIN BRUCE – Investigating Fibre Degradation in the Infant Gut Microbiota ; DIRK HACKENBERGER – Was World War 2 Foundational to the Antimicrobial Resistance Crisis?
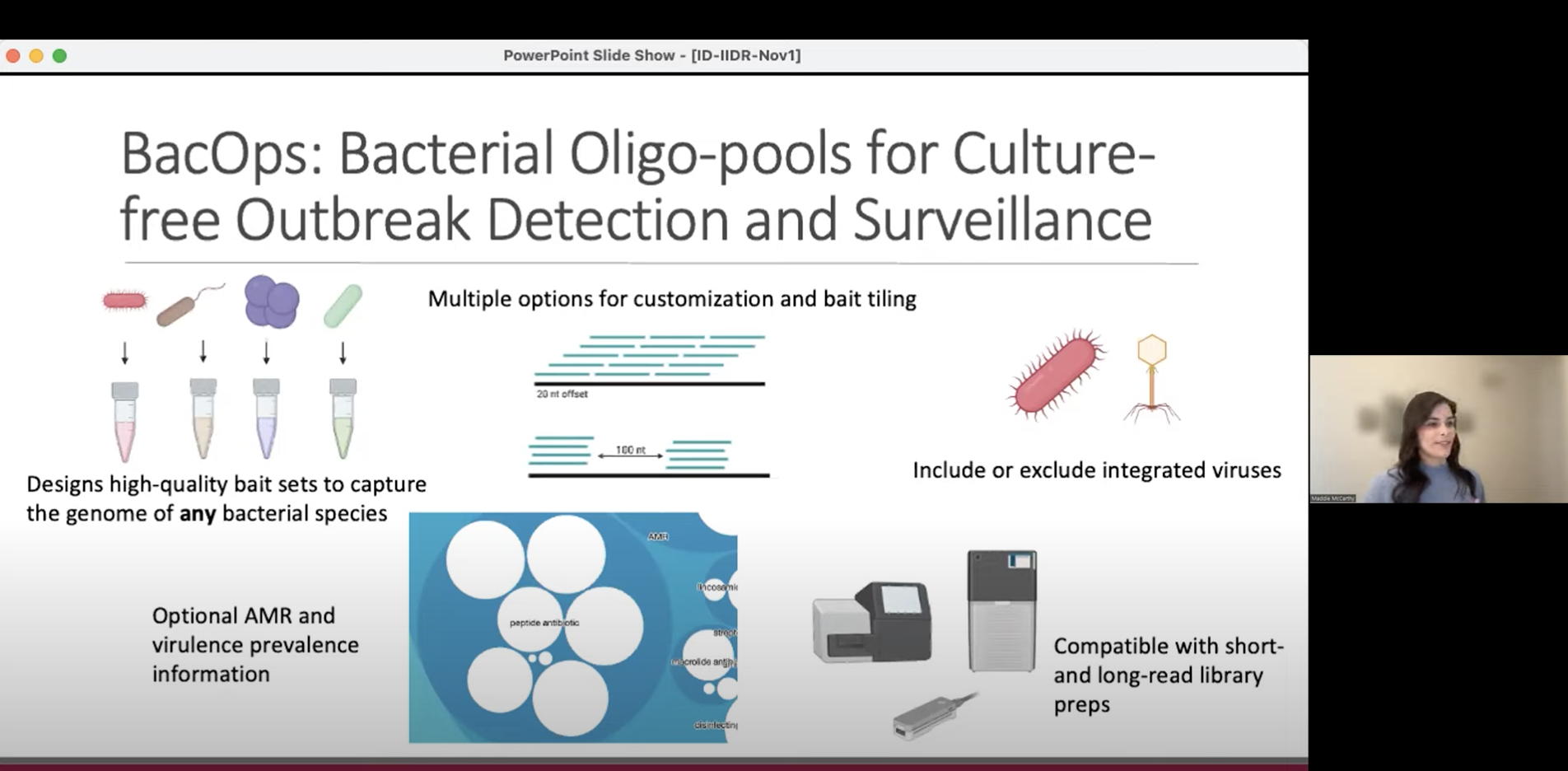
ID-IIDR Rounds – Clostridioides (Clostridium) difficile: new insights from the clinic and laboratory
This one-hour webinar features talks from Marek Smieja (Professor, Pathology & Molecular Medicine), Sheridan Baker (Postdoctoral Fellow, The McArthur Lab and Smieja Lab), and Maddie

2023-2024 Thesis & Inquiry Course Students Kriti Goel (3rd year Biochemistry) – BIOCHEM 3R06 Tiffany Ta (4th year Biomedical Discovery & Commercialization) – BIOMEDDC 4A15 Kristine

The McArthur lab welcomes Brody Duncan, M.D. (Hamilton Health Sciences) as he starts his M.Sc. studies with us, investigating standards and methods for clinical reporting

Baker, S.J.C., J. Maciejewski, M.-T. Usuanlele, J. Gilchrist, D.R. Sharma, D. Bulir, M. Smieja, M. Loeb, M.G. Surette, A.G. McArthur, & D. Mertz. 2023. Investigating

Jacob et al. Emerg Infect Dis. 2023 Jun 12;29(7). Isolating and characterizing emerging SARS-CoV-2 variants is key to understanding virus pathogenesis. In this study, we

While many know of Amogelang (Amos) Raphenya as the Lead CARD Developer and a local, national, and international resource for genomic surveillance of antimicrobial resistance,

Colin Bruce joins us by way of the laboratory of Dr. Jennifer Stearns and will be completing the last year of his PhD studies with

Today the Comprehensive Antibiotic Resistance Database turns 10 years old! While it started a few years before this in the laboratory of Dr. Gerry Wright,

CARD and CZ ID are thrilled to launch a new CZ ID module that allows researchers to detect and analyze antimicrobial resistance (AMR) genes in

The SARS-CoV-2 Illumina GeNome Assembly Line (SIGNAL) has been updated to version 1.6.2, with an important hotfix to address software version incompatibility for rules using