
Day, E.A., L.K. Townsend, S. Rehal, B. Batchuluun, D. Wang, M.R. Morrow, R. Lu, L. Lundenberg, J.H. Lu, E.M. Desjardins, A.R. Raphenya, A.G. McArthur, M.D.
Author: agmcarthur

Keaton W Smith, Brian P Alcock, Shawn French, Maya A Farha, Amogelang R Raphenya, Eric D Brown, & Andrew G McArthur. Microbiol Spectrum. 2023 Nov

antibiotic resistance big data bioinformatics databases epidemiology genomics IIDR lab members molecular epidemiology ontologies presentations software text mining
IIDR Trainee Day 2023
COLIN BRUCE – Investigating Fibre Degradation in the Infant Gut Microbiota ; DIRK HACKENBERGER – Was World War 2 Foundational to the Antimicrobial Resistance Crisis?
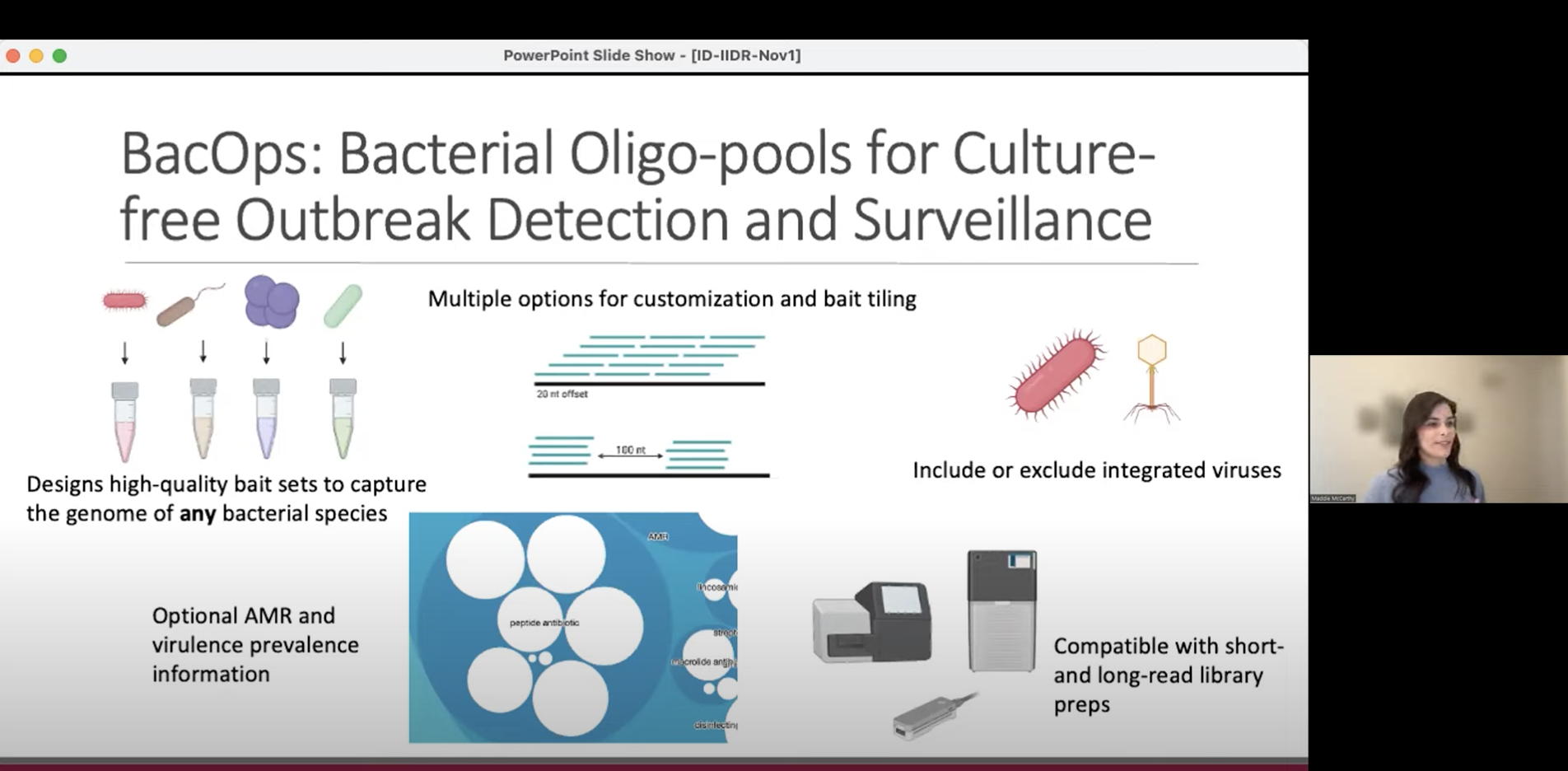
ID-IIDR Rounds – Clostridioides (Clostridium) difficile: new insights from the clinic and laboratory
This one-hour webinar features talks from Marek Smieja (Professor, Pathology & Molecular Medicine), Sheridan Baker (Postdoctoral Fellow, The McArthur Lab and Smieja Lab), and Maddie

2023-2024 Thesis & Inquiry Course Students Kriti Goel (3rd year Biochemistry) – BIOCHEM 3R06 Tiffany Ta (4th year Biomedical Discovery & Commercialization) – BIOMEDDC 4A15 Kristine

The McArthur lab welcomes Brody Duncan, M.D. (Hamilton Health Sciences) as he starts his M.Sc. studies with us, investigating standards and methods for clinical reporting

Amogelang Raphenya makes his triumphant return to the top of the leaderboard!

Baker, S.J.C., J. Maciejewski, M.-T. Usuanlele, J. Gilchrist, D.R. Sharma, D. Bulir, M. Smieja, M. Loeb, M.G. Surette, A.G. McArthur, & D. Mertz. 2023. Investigating

Jacob et al. Emerg Infect Dis. 2023 Jun 12;29(7). Isolating and characterizing emerging SARS-CoV-2 variants is key to understanding virus pathogenesis. In this study, we

While many know of Amogelang (Amos) Raphenya as the Lead CARD Developer and a local, national, and international resource for genomic surveillance of antimicrobial resistance,

Colin Bruce joins us by way of the laboratory of Dr. Jennifer Stearns and will be completing the last year of his PhD studies with

Today the Comprehensive Antibiotic Resistance Database turns 10 years old! While it started a few years before this in the laboratory of Dr. Gerry Wright,


CARD and CZ ID are thrilled to launch a new CZ ID module that allows researchers to detect and analyze antimicrobial resistance (AMR) genes in

The SARS-CoV-2 Illumina GeNome Assembly Line (SIGNAL) has been updated to version 1.6.2, with an important hotfix to address software version incompatibility for rules using

Tsang et al. Microbiol Spectr. 2023 Apr 24;e0190022. Genomic epidemiology can facilitate an understanding of evolutionary history and transmission dynamics of a severe acute respiratory

Curation volunteer Kriti Goel (3rd year Biochemistry) will be joining us as a 2023 Summer Student to sketch out how the Comprehensive Antibiotic Resistance Database
Volunteer starting in 2019, thesis student, summer student, Masters student (2020-2022), and developer, Arman Edalatmand has been a great lab member for over 4 years,

Alcock, B.P., A.R. Raphenya, A. Edalatmand, & A.G. McArthur. 2023. The Comprehensive Antibiotic Resistance Database – curating the global resistome. Oral presentation at the 16th

Arman Edalatmand & Andrew G McArthur. 2023. Database, doi: 10.1093/database/baad023. Scientific literature is published at a rate that makes manual data extraction a highly time-consuming