Category: ontologies

IIDR Trainee Day 2024!
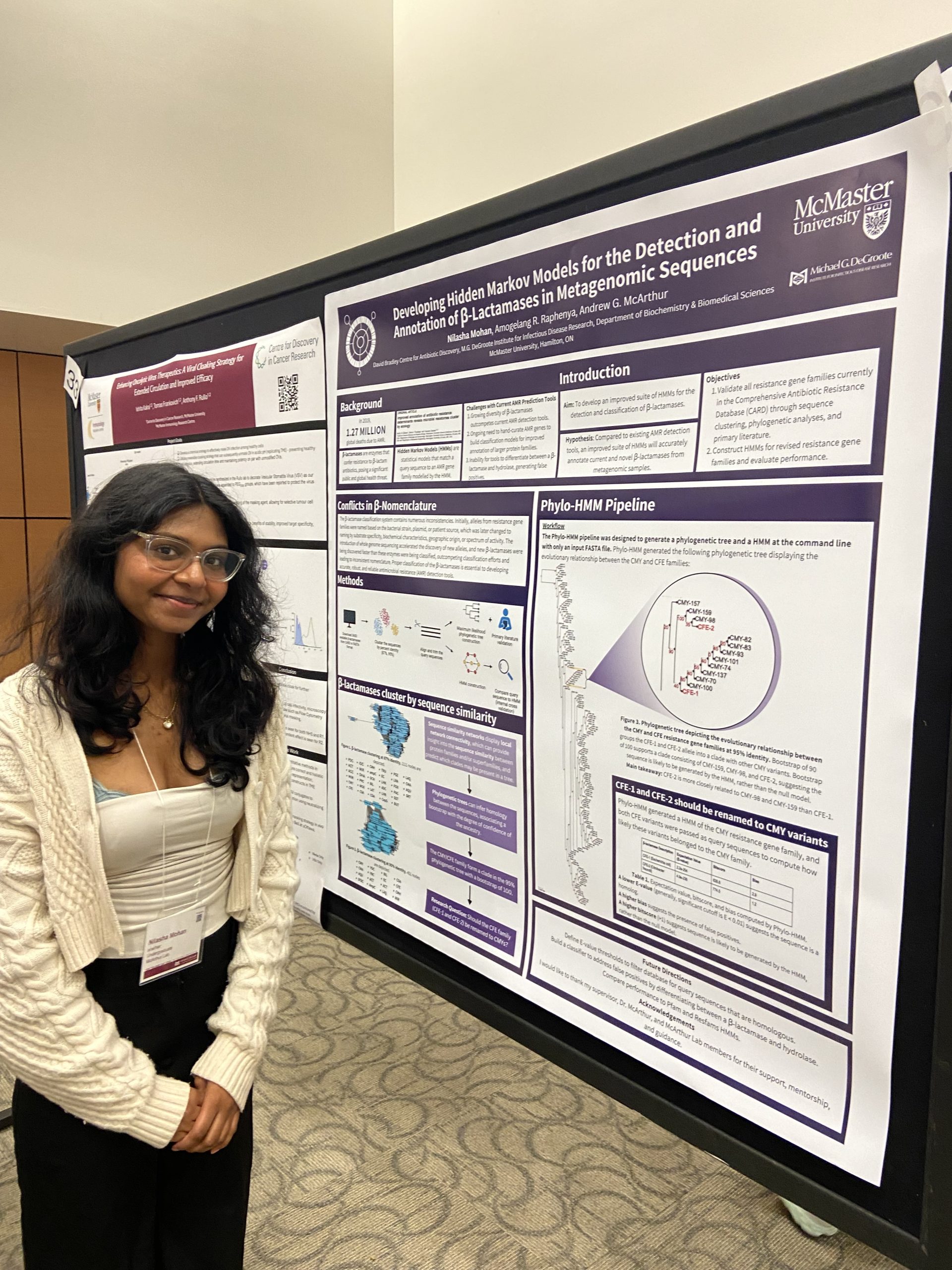
A great day of posters and presentations by the trainees of the Michael G. DeGroote Institute of Infectious Disease Research! Nilasha Mohan, Undergraduate student. Developing

Keaton W Smith, Brian P Alcock, Shawn French, Maya A Farha, Amogelang R Raphenya, Eric D Brown, & Andrew G McArthur. Microbiol Spectrum. 2023 Nov

IIDR Trainee Day 2023
COLIN BRUCE – Investigating Fibre Degradation in the Infant Gut Microbiota ; DIRK HACKENBERGER – Was World War 2 Foundational to the Antimicrobial Resistance Crisis?

While many know of Amogelang (Amos) Raphenya as the Lead CARD Developer and a local, national, and international resource for genomic surveillance of antimicrobial resistance,

Alcock, B.P., A.R. Raphenya, A. Edalatmand, & A.G. McArthur. 2023. The Comprehensive Antibiotic Resistance Database – curating the global resistome. Oral presentation at the 16th

Arman Edalatmand & Andrew G McArthur. 2023. Database, doi: 10.1093/database/baad023. Scientific literature is published at a rate that makes manual data extraction a highly time-consuming

IIDR Trainee Day 2022!
KARYN MUKIRI – Predicting the total resistome KEATON SMITH – Advancements in curation of the Comprehensive Antibiotic Resistance Database JALEES NASIR – Viral fishing expedition:

Alcock et al. 2023. Nucleic Acids Res Oct 20, 2022:gkac920. (online ahead of print) The Comprehensive Antibiotic Resistance Database (CARD; card.mcmaster.ca) combines the Antibiotic Resistance
Emma J. Griffiths et al. Gigascience. 2022 Feb 16;11:giac003. Background: The Public Health Alliance for Genomic Epidemiology (PHA4GE) (https://pha4ge.org) is a global coalition that is

Aaron Petkau performed his Visual & Automated Disease Analytics Program internship (Univ. of Manitoba) with us during the summer of 2020, creating the CARD:Live graphic

New CARD Team Member!
The McArthur lab welcomes new Curator-Developer William Huynh to the Comprehensive Antibiotic Resistance Database staff. Looking forward to pushing CARD forward with William’s help!

Congratulations finishing thesis students!
COVID19 kept us from celebrating together, but we are very proud of the work you have accomplished! Rachel Tran | Biochem 4T15 – Exploring the

CARD 2020: antibiotic resistome surveillance with the Comprehensive Antibiotic Resistance Database
Alcock BP, Raphenya AR, Lau TTY, Tsang KK, Bouchard M, Edalatmand A, Huynh W, Nguyen A-LV, Cheng AA, Liu S, Min SY, Miroshnichenko A, Tran

Speicher, D.J., K. Luinstra, J. Maciejewski, K.K. Tsang, A.G. McArthur, & M. Smieja. 2019. Clostridioides difficile strain divergence over time. Oral presentation at the Association of
The Comprehensive Antibiotic Resistance Database has been updated, http://card.mcmaster.ca CARD Curation: Expanded MCR, OXA & IMP beta-lactamase, and macrolide phosphotransferase (MPH) sequence curation. Updated nomenclature
State of the CARD 2019
A week of lectures, demos, and training for the Comprehensive Antibiotic Resistance Database During McMaster Spring Mid-Term Recess (February 18-24), the McArthur lab is pleased

Antimicrobial Resistance: Emergence, Transmission, and Ecology (ARETE). R. Beiko (PI; Dalhousie University), F. Brinkman (co-PI, Simon Fraser University), A.G. McArthur (co-Applicant) + 4 additional co-Applicants.

Welcome #TeamVirulence, left to right: Rachel Tran (Biochem 3R06), Sally Min (BiomedDC 4A15), Anatoly Miroshnichencko (BiomedDC 4A15), and Rafik El Werfalli (BiomedDC 4A15), who are

The McArthur lab and the Comprehensive Antibiotic Resistance Database are proud to join the Canadian Anti-Infective Innovation Network, International Genomic Epidemiology Application Ontology Consortium, and Integrated Rapid