
Wlodarski, M.A., T.T.Y. Lau, B.P. Alcock, A.R. Raphenya, T.E. Ta, F. Maguire, R.G. Beiko, & A.G. McArthur. 2025. Unmasking antibiotic resistance genes and their pathogen
Category: software

antibiotic resistance bioinformatics genomics lab members molecular epidemiology presentations software text mining
33rd Conference on Intelligent Systems for Molecular Biology
Mukiri, K., B. Alcock, A. Raphenya, & A.G. McArthur. 2025. Mind the gap: predicting the total bacterial resistome in the fight against antimicrobial resistance. Poster

It is with great pleasure that we announce that Dr. Jalees Nasir has been awarded the 2025 McMaster Health Sciences Graduate Student Association (HSGSA) Impact

Lu et al. Genome Med. 2025 May 6;17(1):46. Background: Antimicrobial resistant (AMR) pathogens represent urgent threats to human health, and their surveillance is of paramount

Antibiotics and Resistance: Past, Present, and Future By DIRK L HACKENBERGER, B.Sc. A thesis submitted to the school of graduate studies in partial fulfillment of

bioinformatics COVID19 genomics lab members molecular epidemiology SARS-CoV-2 software virus epidemiology
Congratulations Dr. Jalees Nasir!
Designing Molecular Fishhooks for Virus Survellance Platforms By JALEES A. NASIR, B.Sc A Thesis Submitted to the School of Graduate Studies in partial fulfilment of


Wlodarski, M.A., T.T.Y. Lau, B.P. Alcock, A.R. Raphenya, F. Maguire, R.G. Beiko, T.E. Ta, & A.G. McArthur. 2025. Unmasking antibiotic resistance genes and their pathogen

Hackenberger et al. Appl Environ Microbiol. 2025 Feb 28:e0187624. Better interrogation of antimicrobial resistance requires new approaches to detect the associated genes in metagenomic samples.

Jon Stokes (left) and graduate student Autumn Arnold (right) are excited to launch the ESKAPE Model, an all-new AI tool designed to help the global

Nasir et al. NAR Genomics & Bioinformatics. 2024 Dec 18;6(4):lqae176. The incorporation of sequencing technologies in frontline and public health healthcare settings was vital in
antibiotic resistance bioinformatics databases genomics IIDR lab members molecular epidemiology ontologies presentations software volunteers
IIDR Trainee Day 2024!
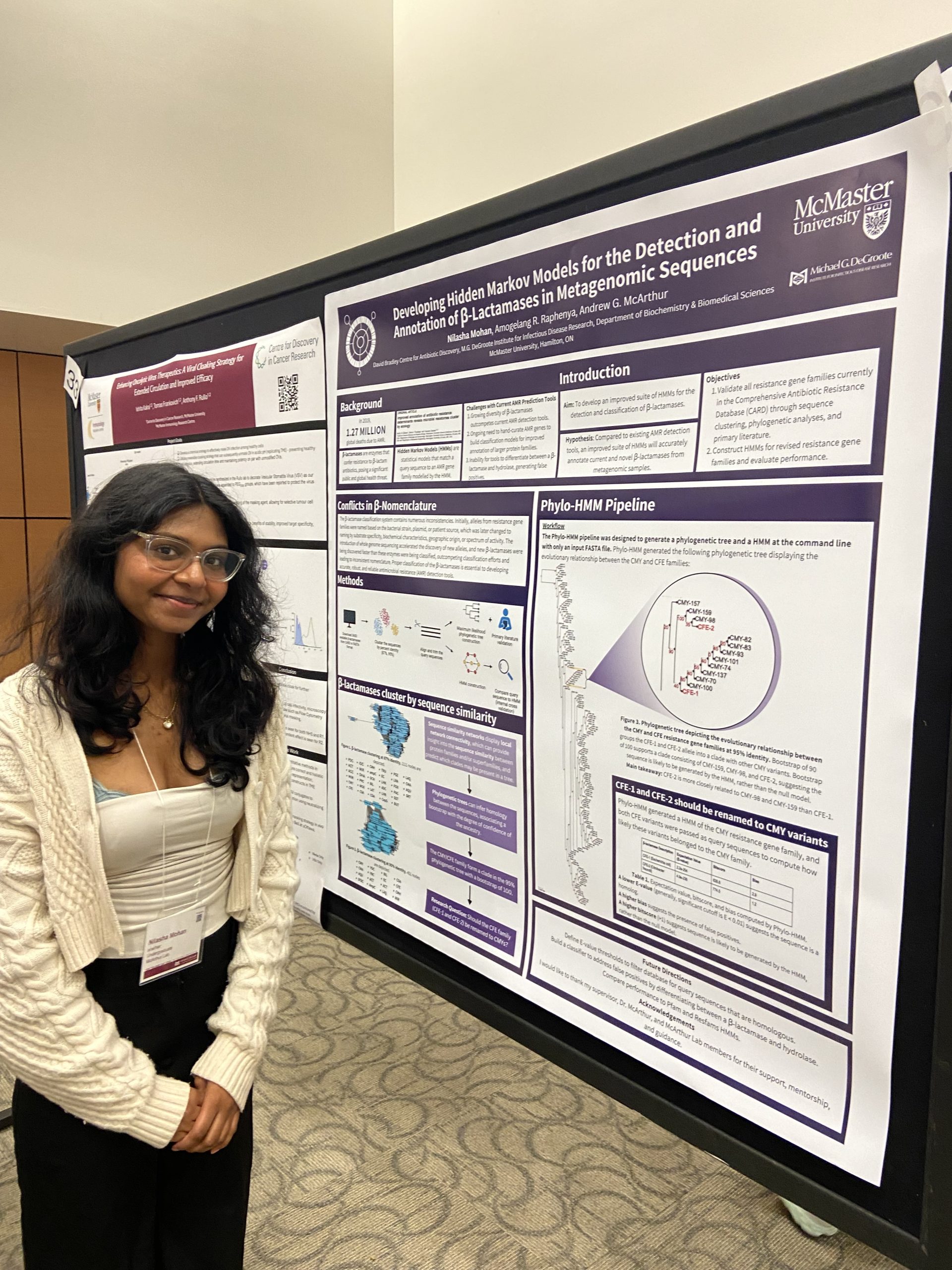
A great day of posters and presentations by the trainees of the Michael G. DeGroote Institute of Infectious Disease Research! Nilasha Mohan, Undergraduate student. Developing
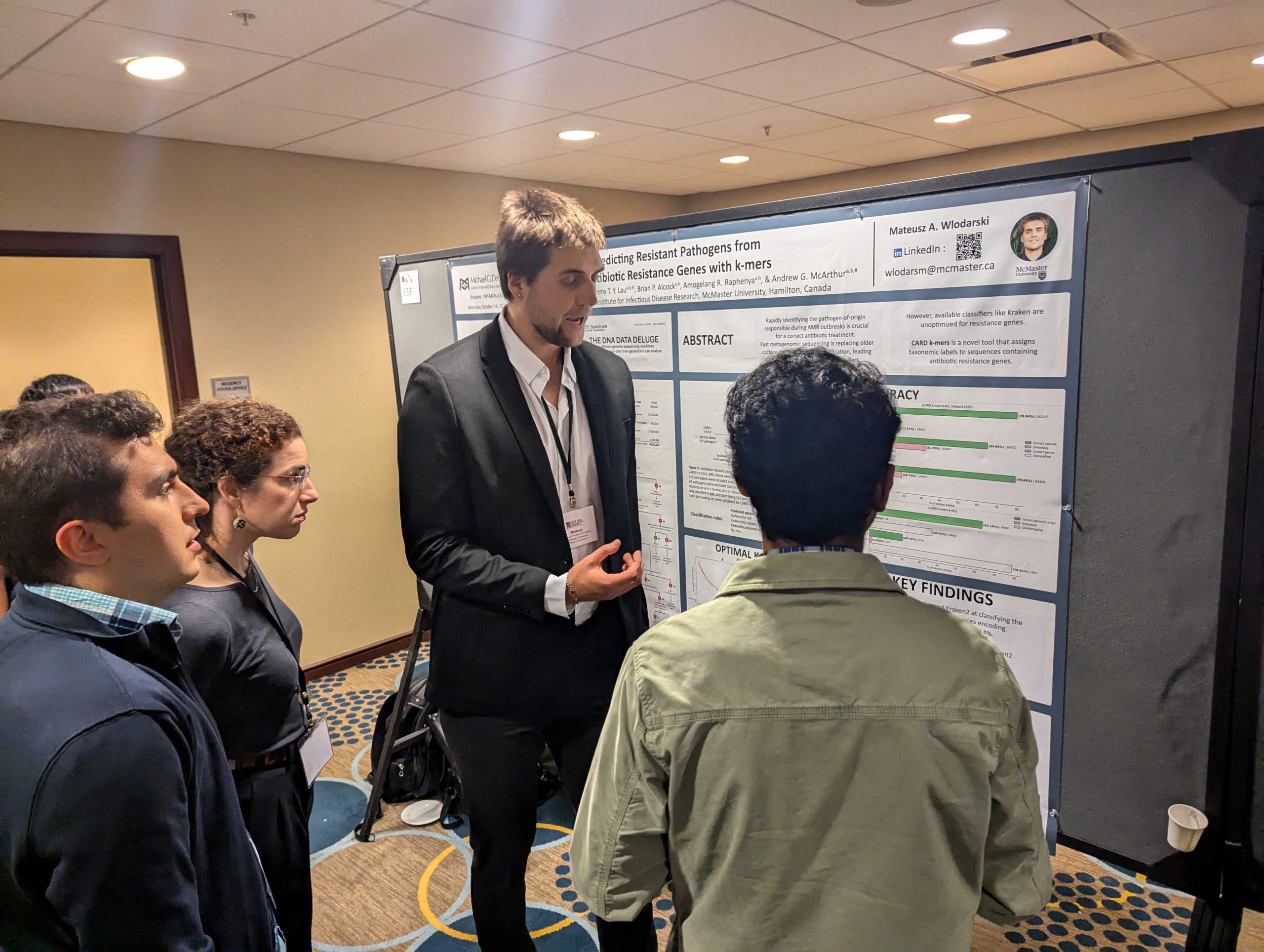
Wlodarski, M.A., T.T.Y. Lau, A.R. Raphenya, B.P. Alcock, & A.G. McArthur. 2024. Accurate pathogen-of-origin classification of antibiotic resistance genes with CARD k-mers. Presentation at the

CSM Mukiri, K.M., B.P. Alcock, & A.G. McArthur. 2024. Increasing the predictive accuracy of the Resistance Gene Identifier by abandoning sole reliance on bitscore.

Gill, E.E., B. Jia, C.L. Murall, R. Poujol, M.Z. Anwar, N.S. John, J. Richardsson, A. Hobb, A.S. Olabode, A. Lepsa, A.T. Duggan, A.D. Tyler, A.

antibiotic resistance big data bioinformatics databases epidemiology genomics IIDR lab members molecular epidemiology ontologies presentations software text mining
IIDR Trainee Day 2023
COLIN BRUCE – Investigating Fibre Degradation in the Infant Gut Microbiota ; DIRK HACKENBERGER – Was World War 2 Foundational to the Antimicrobial Resistance Crisis?

Baker, S.J.C., J. Maciejewski, M.-T. Usuanlele, J. Gilchrist, D.R. Sharma, D. Bulir, M. Smieja, M. Loeb, M.G. Surette, A.G. McArthur, & D. Mertz. 2023. Investigating

Today the Comprehensive Antibiotic Resistance Database turns 10 years old! While it started a few years before this in the laboratory of Dr. Gerry Wright,

CARD and CZ ID are thrilled to launch a new CZ ID module that allows researchers to detect and analyze antimicrobial resistance (AMR) genes in

The SARS-CoV-2 Illumina GeNome Assembly Line (SIGNAL) has been updated to version 1.6.2, with an important hotfix to address software version incompatibility for rules using