
Wlodarski, M.A., T.T.Y. Lau, B.P. Alcock, A.R. Raphenya, T.E. Ta, F. Maguire, R.G. Beiko, & A.G. McArthur. 2025. Unmasking antibiotic resistance genes and their pathogen
Category: antibiotic resistance

Komorowski et al. 2025. Journal of Infectious Diseases, in press. Background: Serratia marcescens is an opportunistic AmpC β-lactamase-producing Enterobacterales associated with intensive care unit outbreaks,

antibiotic resistance bioinformatics genomics lab members molecular epidemiology presentations software text mining
33rd Conference on Intelligent Systems for Molecular Biology
Mukiri, K., B. Alcock, A. Raphenya, & A.G. McArthur. 2025. Mind the gap: predicting the total bacterial resistome in the fight against antimicrobial resistance. Poster

Lu et al. Genome Med. 2025 May 6;17(1):46. Background: Antimicrobial resistant (AMR) pathogens represent urgent threats to human health, and their surveillance is of paramount

Antibiotics and Resistance: Past, Present, and Future By DIRK L HACKENBERGER, B.Sc. A thesis submitted to the school of graduate studies in partial fulfillment of


Wlodarski, M.A., T.T.Y. Lau, B.P. Alcock, A.R. Raphenya, F. Maguire, R.G. Beiko, T.E. Ta, & A.G. McArthur. 2025. Unmasking antibiotic resistance genes and their pathogen

Hackenberger et al. Appl Environ Microbiol. 2025 Feb 28:e0187624. Better interrogation of antimicrobial resistance requires new approaches to detect the associated genes in metagenomic samples.

Two alumni of the McMaster Biochemistry graduate program have joined the McArthur Lab! Dr. Emily Bordeleau got her PhD from McMaster in 2022 under the

Jon Stokes (left) and graduate student Autumn Arnold (right) are excited to launch the ESKAPE Model, an all-new AI tool designed to help the global

Mateusz Wlodarski wins the bioMerieux prize for best poster presentation! Arnold, A., A.R. Raphenya, A.G. McArthur, & J.M. Stokes. 2024. The ESKAPE model: AI-guided antibiotic
antibiotic resistance bioinformatics databases genomics IIDR lab members molecular epidemiology ontologies presentations software volunteers
IIDR Trainee Day 2024!
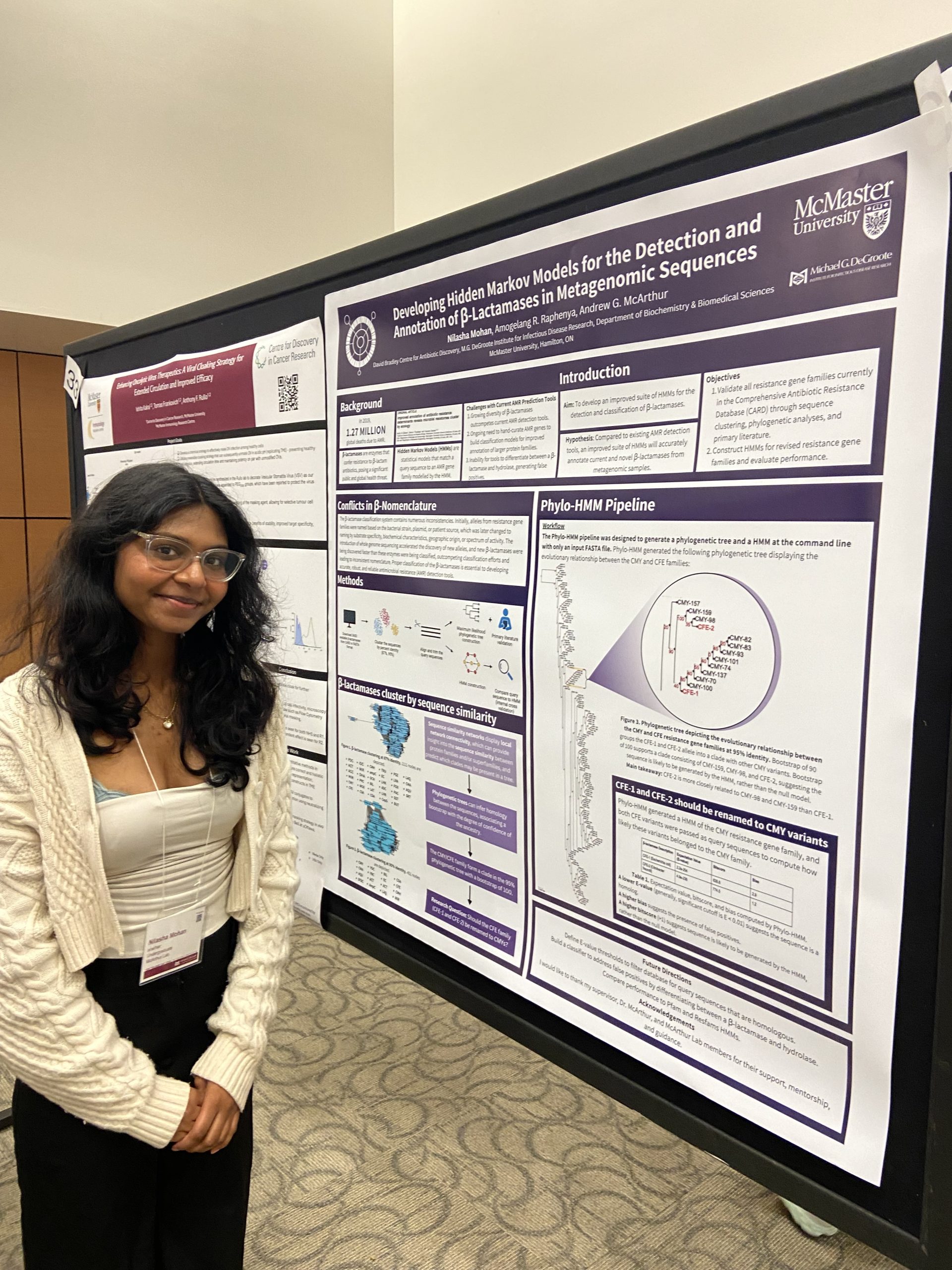
A great day of posters and presentations by the trainees of the Michael G. DeGroote Institute of Infectious Disease Research! Nilasha Mohan, Undergraduate student. Developing
Tsang et al. Microb Genom. 2024 Oct;10(10). Interpreting the phenotypes of bla SHV alleles in Klebsiella pneumoniae genomes is complex. Whilst all strains are expected
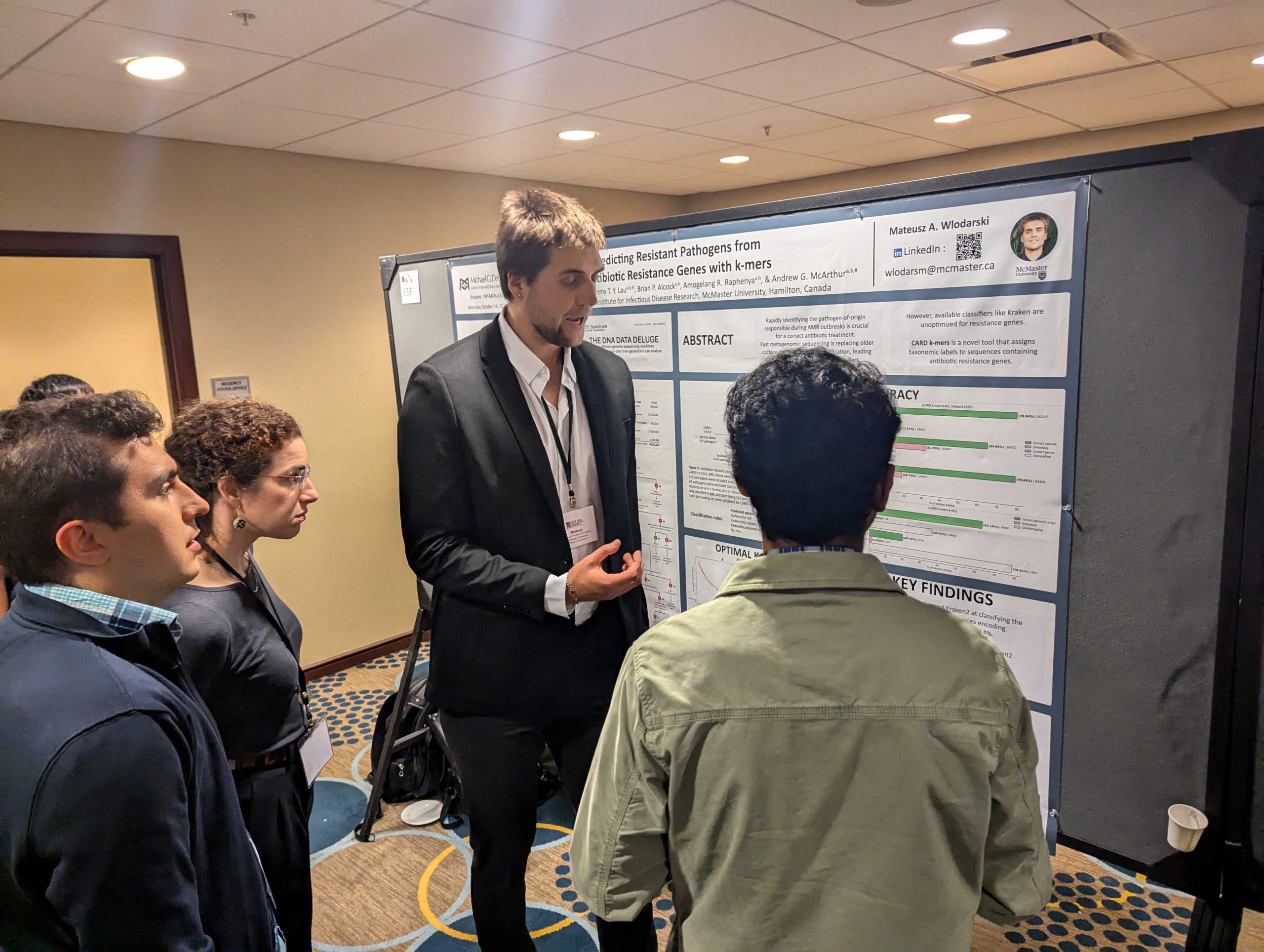
Wlodarski, M.A., T.T.Y. Lau, A.R. Raphenya, B.P. Alcock, & A.G. McArthur. 2024. Accurate pathogen-of-origin classification of antibiotic resistance genes with CARD k-mers. Presentation at the

This one-hour webinar, entitled “DNA sequencing in infectious disease surveillance, diagnosis, and management,” features talks from Andrew McArthur (Professor, Biochemistry and Biomedical Sciences) and Rubayet

CSM Mukiri, K.M., B.P. Alcock, & A.G. McArthur. 2024. Increasing the predictive accuracy of the Resistance Gene Identifier by abandoning sole reliance on bitscore.

Gill, E.E., B. Jia, C.L. Murall, R. Poujol, M.Z. Anwar, N.S. John, J. Richardsson, A. Hobb, A.S. Olabode, A. Lepsa, A.T. Duggan, A.D. Tyler, A.

Alcock, B.P., E.A. Bordeleau, & A.G McArthur. 2024. Improving aminoglycoside resistance surveillance and stewardship efforts through nomenclature harmonization with the Comprehensive Antibiotic Resistance Database. Presentation

antibiotic resistance big data bioinformatics databases genomics molecular epidemiology presentations
Antimicrobial Resistance – Genomes, Big Data and Emerging Technologies – Wellcome Trust, UK
Mukiri, K.M., B.P. Alcock, & A.G. McArthur. 2024. Increasing the predictive accuracy of the Resistance Gene Identifier by abandoning sole reliance on bitscore. Ta, T.E.,

Allison K Guitor, Anna Katyukhina, Margaret Mokomane, Kwana Lechiile, David M Goldfarb, Gerard D Wright, Andrew G McArthur, & Jeffrey M Pernica J Infect Dis.