
Lu et al. Genome Med. 2025 May 6;17(1):46. Background: Antimicrobial resistant (AMR) pathogens represent urgent threats to human health, and their surveillance is of paramount
Category: databases
antibiotic resistance bioinformatics databases genomics IIDR lab members molecular epidemiology ontologies presentations software volunteers
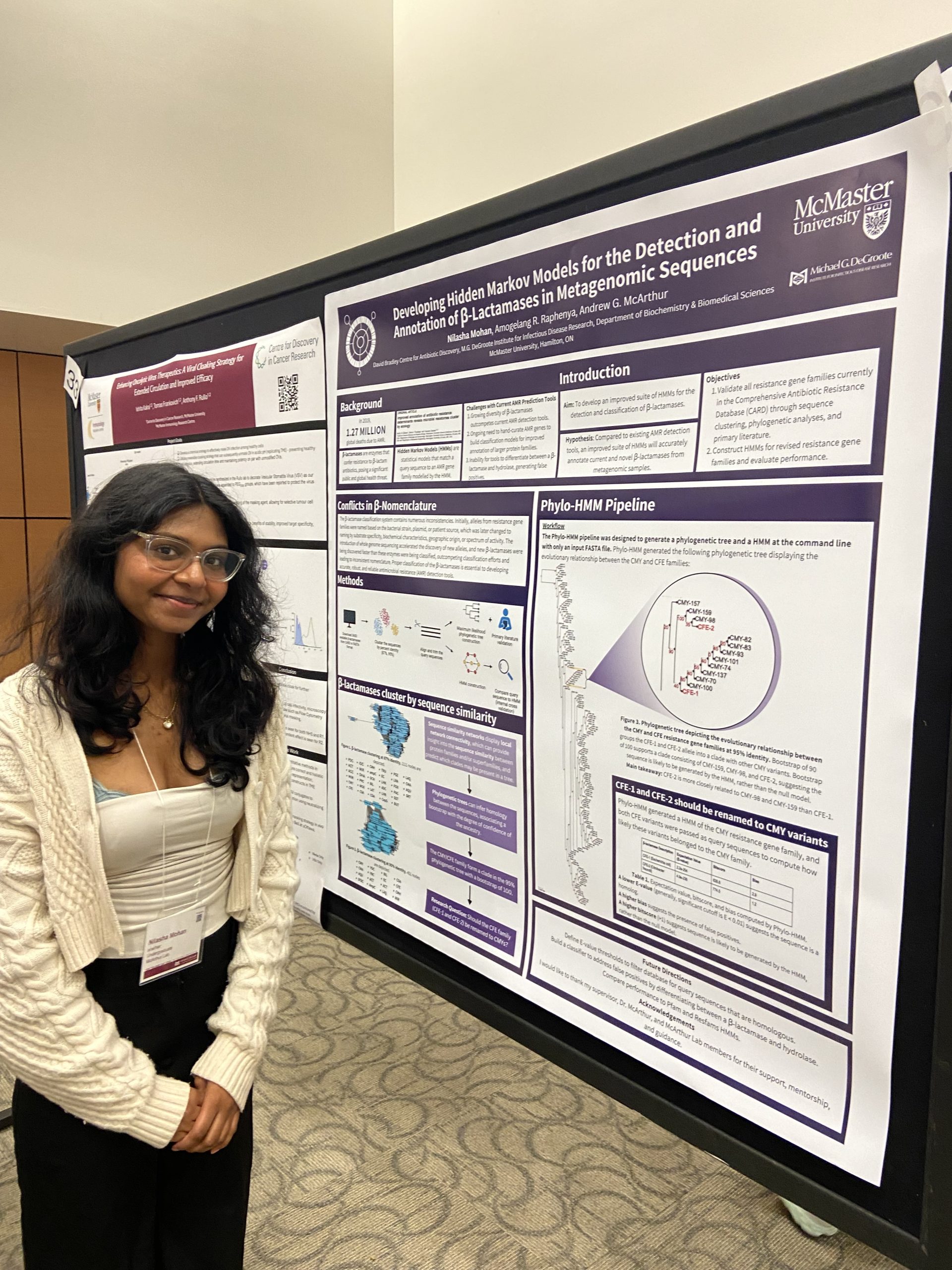
IIDR Trainee Day 2024!
A great day of posters and presentations by the trainees of the Michael G. DeGroote Institute of Infectious Disease Research! Nilasha Mohan, Undergraduate student. Developing

Gill et al. 2024. The Canadian VirusSeq Data Portal & Duotang: open resources for SARS-CoV-2 viral sequences and genomic epidemiology. Microb Genom. 2024 Oct;10(10):001293. The

CSM Mukiri, K.M., B.P. Alcock, & A.G. McArthur. 2024. Increasing the predictive accuracy of the Resistance Gene Identifier by abandoning sole reliance on bitscore.

Gill, E.E., B. Jia, C.L. Murall, R. Poujol, M.Z. Anwar, N.S. John, J. Richardsson, A. Hobb, A.S. Olabode, A. Lepsa, A.T. Duggan, A.D. Tyler, A.

Alcock, B.P., E.A. Bordeleau, & A.G McArthur. 2024. Improving aminoglycoside resistance surveillance and stewardship efforts through nomenclature harmonization with the Comprehensive Antibiotic Resistance Database. Presentation

antibiotic resistance big data bioinformatics databases genomics molecular epidemiology presentations
Antimicrobial Resistance – Genomes, Big Data and Emerging Technologies – Wellcome Trust, UK
Mukiri, K.M., B.P. Alcock, & A.G. McArthur. 2024. Increasing the predictive accuracy of the Resistance Gene Identifier by abandoning sole reliance on bitscore. Ta, T.E.,

Keaton W Smith, Brian P Alcock, Shawn French, Maya A Farha, Amogelang R Raphenya, Eric D Brown, & Andrew G McArthur. Microbiol Spectrum. 2023 Nov

antibiotic resistance big data bioinformatics databases epidemiology genomics IIDR lab members molecular epidemiology ontologies presentations software text mining
IIDR Trainee Day 2023
COLIN BRUCE – Investigating Fibre Degradation in the Infant Gut Microbiota ; DIRK HACKENBERGER – Was World War 2 Foundational to the Antimicrobial Resistance Crisis?

While many know of Amogelang (Amos) Raphenya as the Lead CARD Developer and a local, national, and international resource for genomic surveillance of antimicrobial resistance,

Today the Comprehensive Antibiotic Resistance Database turns 10 years old! While it started a few years before this in the laboratory of Dr. Gerry Wright,

CARD and CZ ID are thrilled to launch a new CZ ID module that allows researchers to detect and analyze antimicrobial resistance (AMR) genes in

Alcock, B.P., A.R. Raphenya, A. Edalatmand, & A.G. McArthur. 2023. The Comprehensive Antibiotic Resistance Database – curating the global resistome. Oral presentation at the 16th

Arman Edalatmand & Andrew G McArthur. 2023. Database, doi: 10.1093/database/baad023. Scientific literature is published at a rate that makes manual data extraction a highly time-consuming
antibiotic resistance bioinformatics databases genomics lab members molecular epidemiology outreach teaching
CBW & VTEC 2023
They’re back! Thanks to Karyn Mukiri, Jalees Nasir, and Madeline McCarthy for serving as Teaching Assistants for the re-booted Canadian Bioinformatics Workshop in Infectious Disease

The McArthur Lab welcomes Team Meta-Model, a volunteer curation team led by former BioPharm Co-Op Keaton Smith, working on curating and upgrading the Comprehensive Antibiotic

antibiotic resistance awards bioinformatics Co-Op positions databases epidemiology genomics IIDR lab members ontologies presentations
IIDR Trainee Day 2022!
KARYN MUKIRI – Predicting the total resistome KEATON SMITH – Advancements in curation of the Comprehensive Antibiotic Resistance Database JALEES NASIR – Viral fishing expedition:

Alcock et al. 2023. Nucleic Acids Res Oct 20, 2022:gkac920. (online ahead of print) The Comprehensive Antibiotic Resistance Database (CARD; card.mcmaster.ca) combines the Antibiotic Resistance

antibiotic resistance bioinformatics databases genomics molecular epidemiology publications software
Enabling genomic island prediction and comparison in multiple genomes to investigate bacterial evolution and outbreaks
Bertelli et al. Microb Genom. 2022 May;8(5). doi: 10.1099/mgen.0.000818. Outbreaks of virulent and/or drug-resistant bacteria have a significant impact on human health and major economic

Today we say goodbye to William Huynh, who first joined us as an undergraduate thesis student and then stayed on to work as the Comprehensive