
Jon Stokes (left) and graduate student Autumn Arnold (right) are excited to launch the ESKAPE Model, an all-new AI tool designed to help the global

Nasir et al. NAR Genomics & Bioinformatics. 2024 Dec 18;6(4):lqae176. The incorporation of sequencing technologies in frontline and public health healthcare settings was vital in

Mateusz Wlodarski wins the bioMerieux prize for best poster presentation! Arnold, A., A.R. Raphenya, A.G. McArthur, & J.M. Stokes. 2024. The ESKAPE model: AI-guided antibiotic
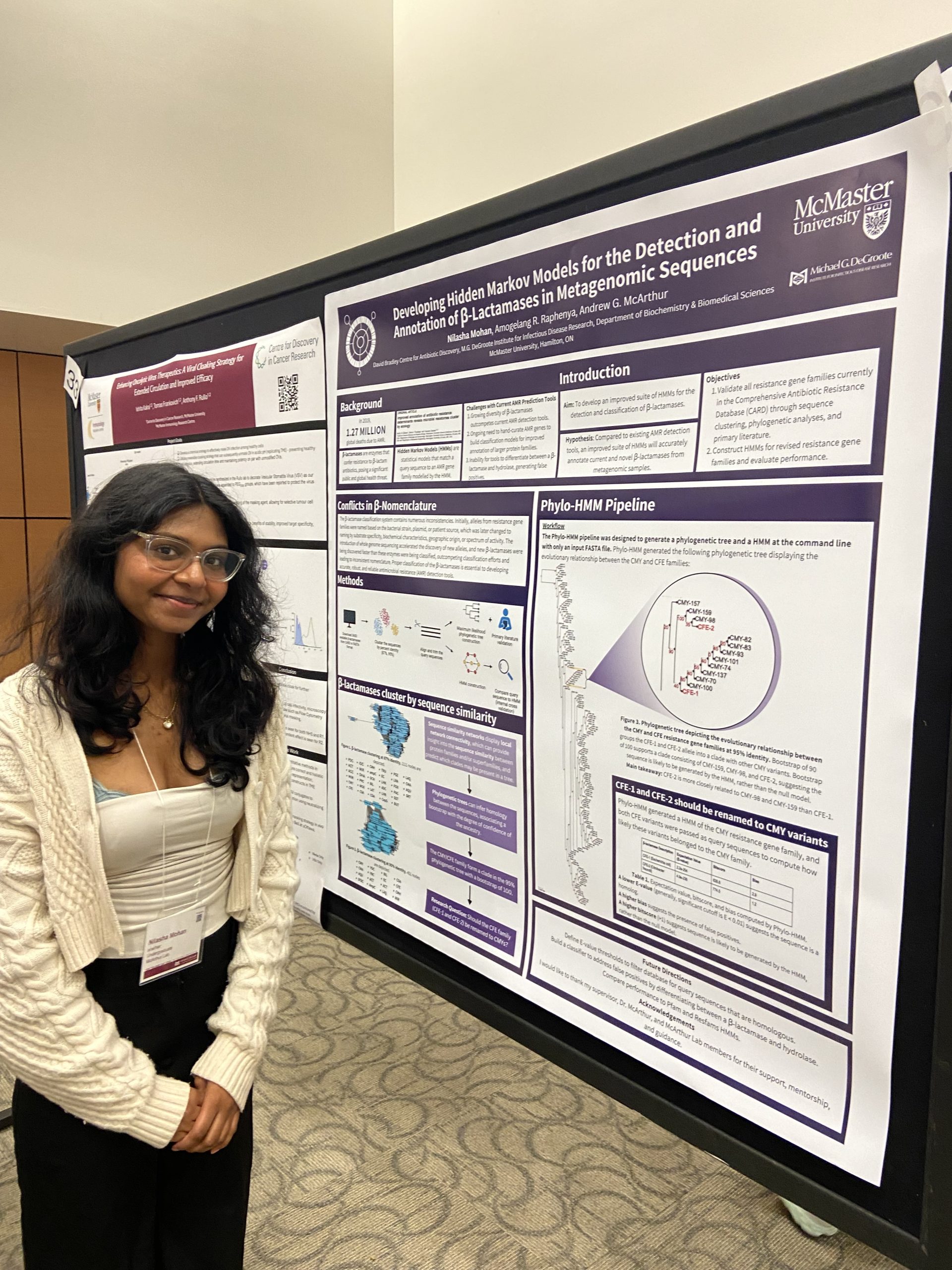
antibiotic resistance bioinformatics databases genomics IIDR lab members molecular epidemiology ontologies presentations software volunteers
IIDR Trainee Day 2024!
A great day of posters and presentations by the trainees of the Michael G. DeGroote Institute of Infectious Disease Research! Nilasha Mohan, Undergraduate student. Developing
Tsang et al. Microb Genom. 2024 Oct;10(10). Interpreting the phenotypes of bla SHV alleles in Klebsiella pneumoniae genomes is complex. Whilst all strains are expected

Gill et al. 2024. The Canadian VirusSeq Data Portal & Duotang: open resources for SARS-CoV-2 viral sequences and genomic epidemiology. Microb Genom. 2024 Oct;10(10):001293. The
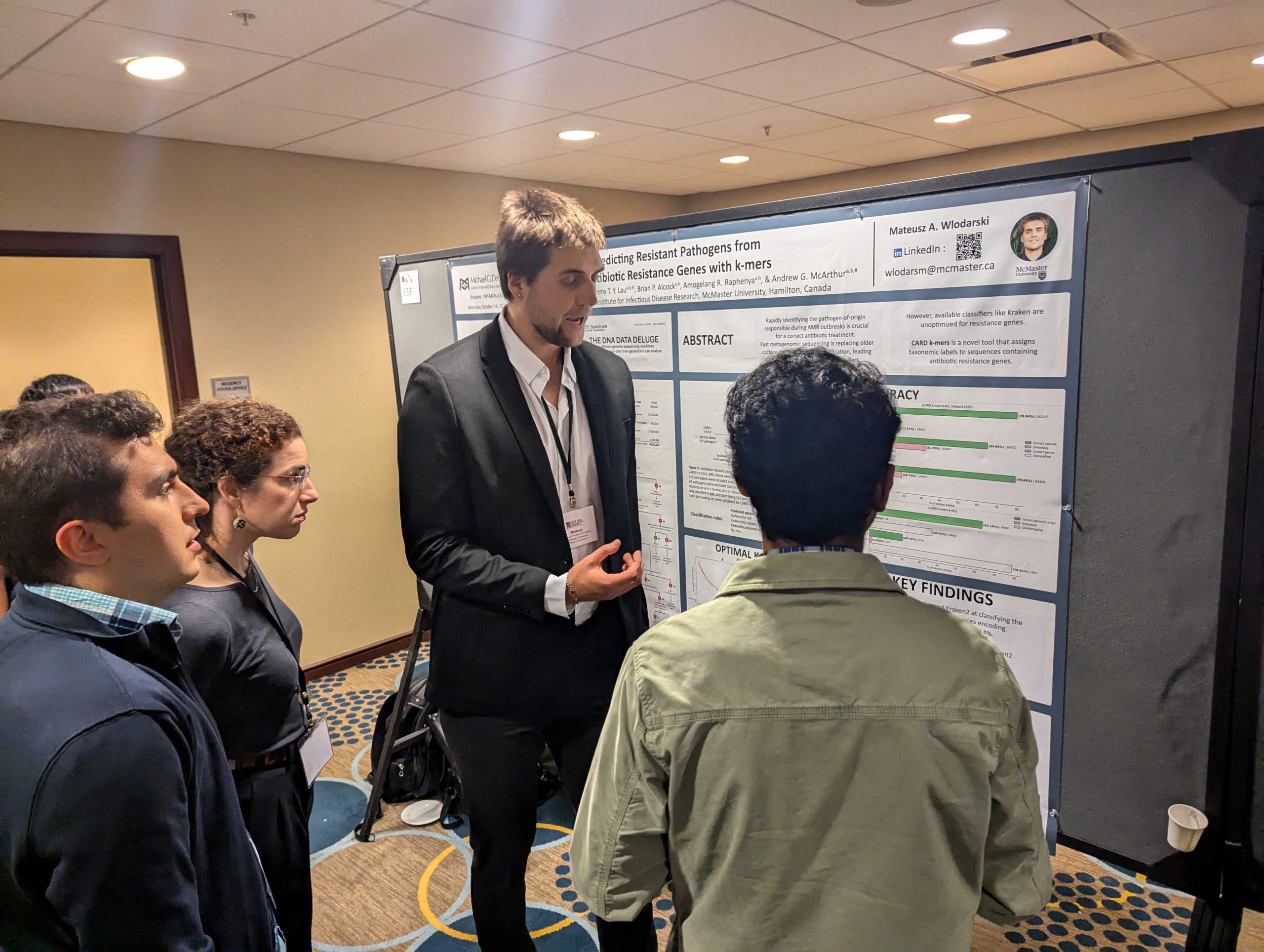
Wlodarski, M.A., T.T.Y. Lau, A.R. Raphenya, B.P. Alcock, & A.G. McArthur. 2024. Accurate pathogen-of-origin classification of antibiotic resistance genes with CARD k-mers. Presentation at the

This one-hour webinar, entitled “DNA sequencing in infectious disease surveillance, diagnosis, and management,” features talks from Andrew McArthur (Professor, Biochemistry and Biomedical Sciences) and Rubayet

71 undergraduates, 9 MSc, 16 MBDC, 6 PhD, 2 PDF, and 12 staff, co-ops, and summer students! And 50+ publications!

This September we welcome new graduate students Tiffany Ta and Suhayb Hafiz. Tiffany joins us from the McMaster Biomedical Discovery & Commercialization program and will

Welcome to the lab thesis students Trisha Parchoor, Sidrah Shaikh, Nilasha Mohan, & Abby Williams!

Lab alumni Arman Edalatmand returned and won the match!

CSM Mukiri, K.M., B.P. Alcock, & A.G. McArthur. 2024. Increasing the predictive accuracy of the Resistance Gene Identifier by abandoning sole reliance on bitscore.

Gill, E.E., B. Jia, C.L. Murall, R. Poujol, M.Z. Anwar, N.S. John, J. Richardsson, A. Hobb, A.S. Olabode, A. Lepsa, A.T. Duggan, A.D. Tyler, A.

Welcome to BDC post-grad Tiffany Ta, and incoming thesis students Nilasha Mohan and Sidrah Shaikh to their summer placements!

Alcock, B.P., E.A. Bordeleau, & A.G McArthur. 2024. Improving aminoglycoside resistance surveillance and stewardship efforts through nomenclature harmonization with the Comprehensive Antibiotic Resistance Database. Presentation

antibiotic resistance big data bioinformatics databases genomics molecular epidemiology presentations
Antimicrobial Resistance – Genomes, Big Data and Emerging Technologies – Wellcome Trust, UK
Mukiri, K.M., B.P. Alcock, & A.G. McArthur. 2024. Increasing the predictive accuracy of the Resistance Gene Identifier by abandoning sole reliance on bitscore. Ta, T.E.,

Congratulations to Colin Bruce for winning First Place PhD poster at the Biochemistry & Biomedical Sciences Research Symposium!

Allison K Guitor, Anna Katyukhina, Margaret Mokomane, Kwana Lechiile, David M Goldfarb, Gerard D Wright, Andrew G McArthur, & Jeffrey M Pernica J Infect Dis.
clinical metadata COVID19 genomics molecular epidemiology publications SARS-CoV-2 virus epidemiology
Chronic COVID-19 infection in an immunosuppressed patient shows changes in lineage over time: a case report
Sheridan J C Baker , Landry E Nfonsam , Daniela Leto , Candy Rutherford , Marek Smieja , & Andrew G McArthur Virol J. 2024